
Enol



In organic chemistry, enols are a type of Functional group or intermediate in organic chemistry containing a group with the formula C=C(OH) (R = many substituents). The term enol is an abbreviation of alkenol, a portmanteau deriving from "-ene"/"alkene" and the "-ol". Many kinds of enols are known.[1]
Keto–enol tautomerism refers to a chemical equilibrium between a "keto" form (a carbonyl, named for the common ketone case) and an enol. The interconversion of the two forms involves the transfer of an alpha hydrogen atom and the reorganisation of bonding electrons. The keto and enol forms are tautomers of each other.[2]
Enolization
Organic esters, ketones, and aldehydes with an α-hydrogen (C−H bond adjacent to the carbonyl group) often form enols. The reaction involves migration of a proton (H) from carbon to oxygen:[1]
- RC(=O)CHR′R′′ ⇌ RC(OH)=CR′R′′
In the case of ketones, the conversion is called a keto-enol tautomerism, although this name is often more generally applied to all such tautomerizations. Usually the equilibrium constant is so small that the enol is undetectable spectroscopically.
In some compounds with two (or more) carbonyls, the enol form becomes dominant. The behavior of 2,4-pentanedione illustrates this effect:[3]
| carbonyl | enol | Kenolization |
|---|---|---|
| Acetaldehyde CH3CHO |
CH2=CHOH | 5.8×10−7 |
| Acetone CH3C(O)CH3 |
CH3C(OH)=CH2 | 5.12×10−7 |
| Methyl acetate CH3CO2CH3 |
CH2=CH(OH)OCH3 | 4×10−20 |
| Acetophenone C6H5C(O)CH3 |
C6H5C(OH)=CH2 | 1×10−8 |
| Acetylacetone CH3C(O)CH2C(O)CH3 |
CH3C(O)CH=C(OH)CH3 | 0.27 |
| Trifluoroacetylacetone CH3C(O)CH2C(O)CF3 |
CH3C(O)CH=C(OH)CF3 | 32 |
| Hexafluoroacetylacetone CF3C(O)CH2C(O)CF3 |
CF3C(O)CH=C(OH)CF3 | ~104 |
| Cyclohexa-2,4-dienone | Phenol C6H5OH |
>1012 |
Enols are derivatives of vinyl alcohol, with a C=C−OH connectivity. Deprotonation of organic carbonyls gives the enolate anion, which are a strong nucleophile. A classic example for favoring the keto form can be seen in the equilibrium between vinyl alcohol and acetaldehyde (K = [enol]/[keto] ≈ 3×10−7). In 1,3-diketones, such as acetylacetone (2,4-pentanedione), the enol form is favored.
The acid-catalyzed conversion of an enol to the keto form proceeds by proton transfer from O to carbon. The process does not occur intramolecularly, but requires participation of solvent or other mediators.
Stereochemistry of ketonization
If R1 and R2 (note equation at top of page) are different substituents, there is a new stereocenter formed at the alpha position when an enol converts to its keto form. Depending on the nature of the three R groups, the resulting products in this situation would be diastereomers or enantiomers.[citation needed]
Enediols
Enediols are alkenes with a hydroxyl group on each carbon of the C=C double bond. Normally such compounds are disfavored components in equilibria with acyloins. One special case is catechol, where the C=C subunit is part of an aromatic ring. In some other cases however, enediols are stabilized by flanking carbonyl groups. These stabilized enediols are called reductones. Such species are important in glycochemistry, e.g., the Lobry de Bruyn-van Ekenstein transformation.[5]
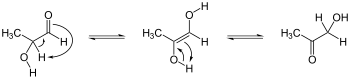
Keto-enediol tautomerizations. Enediol in the center; acyloin isomers at left and right. Ex. is hydroxyacetone, shown at right.


Ribulose-1,5-bisphosphate is a key substrate in the Calvin cycle of photosynthesis. In the Calvin cycle, the ribulose equilibrates with the enediol, which then binds carbon dioxide. The same enediol is also susceptible to attack by oxygen (O2) in the (undesirable) process called photorespiration.

Phenols
Phenols represent a kind of enol. For some phenols and related compounds, the keto tautomer plays an important role. Many of the reactions of resorcinol involve the keto tautomer, for example. Naphthalene-1,4-diol exists in observable equilibrium with the diketone tetrahydronaphthalene-1,4-dione.[6]

Biochemistry
Keto–enol tautomerism is important in several areas of biochemistry.[citation needed]
The high phosphate-transfer potential of phosphoenolpyruvate results from the fact that the phosphorylated compound is "trapped" in the less thermodynamically favorable enol form, whereas after dephosphorylation it can assume the keto form.[citation needed]
The enzyme enolase catalyzes the dehydration of 2-phosphoglyceric acid to the enol phosphate ester. Metabolism of PEP to pyruvic acid by pyruvate kinase (PK) generates adenosine triphosphate (ATP) via substrate-level phosphorylation.[7]

|

|

| ||||
| H2O | ADP | ATP | ||||
| H2O | ||||||
Reactivity
Addition of electrophiles
The terminus of the double bond in enols is nucleophilic. Its reactions with electrophilic organic compounds is important in biochemistry as well as synthetic organic chemistry. In the former area, the fixation of carbon dioxide involves addition of CO2 to an enol.[citation needed]
Deprotonation: enolates
Deprotonation of enolizable ketones, aldehydes, and esters gives enolates.[8][9] Enolates can be trapped by the addition of electrophiles at oxygen. Silylation gives silyl enol ether.[10] Acylation gives esters such as vinyl acetate.[11]
Stable enols
In general, enols are less stable than their keto equivalents because of the favorability of the C=O double bond over C=C double bond. However, enols can be stabilized kinetically or thermodynamically.[citation needed]
Some enols are sufficiently stabilized kinetically so that they can be characterized.[citation needed]

Delocalization can stabilize the enol tautomer. Thus, very stable enols are phenols.[13] Another stabilizing factor in 1,3-dicarbonyls is intramolecular hydrogen bonding.[14] Both of these factors influence the enol-dione equilibrium in acetylacetone.
See also
- Alkenal
- Enolase
- Ketone
- Ynol
- Geminal diol, another form of ketones and aldehydes in water solutions
- Regioselectivity
References
- ^ a b Smith MB, March J (2001). Advanced Organic Chemistry (5th ed.). New York: Wiley Interscience. pp. 1218–1223. ISBN 0-471-58589-0.
- ^ Clayden, Jonathan; Greeves, Nick; Warren, Stuart (2012). Organic chemistry (2nd ed.). New York: Oxford University Press. pp. 450–451. ISBN 978-0-19-927029-3.
- ^ Manbeck, Kimberly A.; Boaz, Nicholas C.; Bair, Nathaniel C.; Sanders, Allix M. S.; Marsh, Anderson L. (2011). "Substituent Effects on Keto–Enol Equilibria Using NMR Spectroscopy". J. Chem. Educ. 88 (10): 1444–1445. Bibcode:2011JChEd..88.1444M. doi:10.1021/ed1010932.
- ^ Guthrie, J. Peter; Povar, Igor (2013). "Equilibrium constants for enolization in solution by computation alone". Journal of Physical Organic Chemistry. 26 (12): 1077–1083. doi:10.1002/poc.3168.
- ^ Schank, Kurt (1972). "Reductones". Synthesis. 1972 (4): 176–90. doi:10.1055/s-1972-21845. S2CID 260331550.
- ^ Kündig, E. Peter; Enríquez García, Alvaro; Lomberget, Thierry; Bernardinelli, Gérald (2006). "Rediscovery, Isolation, and Asymmetric Reduction of 1,2,3,4-Tetrahydronaphthalene-1,4-dione and Studies of Its [Cr(CO)3] Complex". Angewandte Chemie International Edition. 45 (1): 98–101. doi:10.1002/anie.200502588. PMID 16304647.
- ^ Berg, Jeremy M.; Tymoczko, Stryer (2002). Biochemistry (5th ed.). New York: W.H. Freeman and Company. ISBN 0-7167-3051-0.
- ^ Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, ISBN 978-0-471-72091-1
- ^ Manfred Braun (2015). Modern Enolate Chemistry: From Preparation to Applications in Asymmetric Synthesis. Wiley-VCH. doi:10.1002/9783527671069. ISBN 9783527671069.
- ^ Mukaiyama, T.; Kobayashi, S. Org. React. 1994, 46, 1. doi:10.1002/0471264180.or046.01
- ^ G. Roscher (2007). "Vinyl Esters". Ullmann's Encyclopedia of Chemical Technology. Weinheim: Wiley-VCH. doi:10.1002/14356007.a27_419. ISBN 978-3527306732. S2CID 241676899.
- ^ "Stable simple enols". Journal of the American Chemical Society. 1989. doi:10.1021/ja00203a019.
- ^ Clayden, Jonathan (2012). Organic Chemistry. Oxford University Press. pp. 456–459.
- ^ Zhou, Yu-Qiang; Wang, Nai-Xing; Xing, Yalan; Wang, Yan-Jing; Hong, Xiao-Wei; Zhang, Jia-Xiang; Chen, Dong-Dong; Geng, Jing-Bo; Dang, Yanfeng; Wang, Zhi-Xiang (2013-01-14). "Stable acyclic aliphatic solid enols: synthesis, characterization, X-ray structure analysis and calculations". Scientific Reports. 3 (1): 1058. Bibcode:2013NatSR...3E1058Z. doi:10.1038/srep01058. ISSN 2045-2322. PMC 3544012. PMID 23320139.
External links
| Authority control databases: National |
|---|