
Aza-Cope rearrangement
| Aza-Cope rearrangement | |
|---|---|
| Named after | Arthur C. Cope |
| Reaction type | Rearrangement reaction |
| Identifiers | |
| RSC ontology ID | RXNO:0000197 |
Rearrangements, especially those that can participate in cascade reactions, such as the aza-Cope rearrangements, are of high practical as well as conceptual importance in organic chemistry, due to their ability to quickly build structural complexity out of simple starting materials. The aza-Cope rearrangements are examples of heteroatom versions of the Cope rearrangement, which is a [3,3]-sigmatropic rearrangement that shifts single and double bonds between two allylic components. In accordance with the Woodward-Hoffman rules, thermal aza-Cope rearrangements proceed suprafacially.[1] Aza-Cope rearrangements are generally classified by the position of the nitrogen in the molecule (see figure):
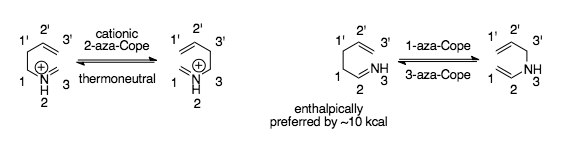
The first example of an aza-Cope rearrangement was the ubiquitous cationic 2-aza-Cope rearrangement, which takes place at temperatures 100-200 °C lower than the Cope rearrangement due to the facile nature of the rearrangement.[2] The facile nature of this rearrangement is attributed both to the fact that the cationic 2-aza-Cope is inherently thermoneutral, meaning there's no bias for the starting material or product, as well as to the presence of the charged heteroatom in the molecule, which lowers the activation barrier. Less common are the 1-aza-Cope rearrangement and the 3-aza-Cope rearrangement, which are the microscopic reverse of each other. The 1- and 3-aza-Cope rearrangements have high activation barriers and limited synthetic applicability, accounting for their relative obscurity.[3][4][5]
To maximize its synthetic utility, the cationic 2-aza-Cope rearrangement is normally paired with a thermodynamic bias toward one side of the rearrangement. The most common and synthetically useful strategy couples the cationic 2-aza-Cope rearrangement with a Mannich cyclization, and is the subject of much of this article. This tandem aza-Cope/Mannich reaction is characterized by its mild reaction conditions, diastereoselectivity, and wide synthetic applicability. It provides easy access to acyl-substituted pyrrolidines, a structure commonly found in natural products such as alkaloids, and has been used in the synthesis of a number of them, notably strychnine and crinine.[6] Larry E. Overman and coworkers have done extensive research on this reaction.[1]
The cationic 2-aza-Cope rearrangement

The cationic 2-aza-Cope rearrangement, most properly called the 2-azonia-[3,3]-sigmatropic rearrangement, has been thoroughly studied by Larry E. Overman and coworkers. It is the most extensively studied of the aza-Cope rearrangements due to the mild conditions required to carry the arrangement out, as well as for its many synthetic applications, notably in alkaloid synthesis. Thermodynamically, the general 2-aza-Cope rearrangement does not have a product bias, as the bonds broken and formed are equivalent in either direction of the reaction, similar to the Cope rearrangement. The presence of the ionic nitrogen heteroatom accounts for the more facile rearrangement of the cationic 2-aza-Cope rearrangement in comparison to the Cope rearrangement. Hence, it is often paired with a thermodynamic sink to bias a rearrangement product.[1]
In 1950, Horowitz and Geissman reported the first example of the 2-aza-Cope rearrangement, a surprising result in a failed attempt to synthesize an amino alcohol.[2] This discovery identified the basic mechanism of the rearrangement, as the product was most likely produced through a nitrogen analog of the Cope rearrangement. Treatment of an allylbenzylamine (A) with formic acid and formaldehyde leads to an amino alcohol (B). The amino alcohol converts to an imine under addition of acid (C), which undergoes the cationic 2-aza-Cope rearrangement (D). Water hydrolyses the iminium ion to an amine (E). Treating this starting material with only formaldehyde showed that alkylation of the amine group occurred after the cationic 2-aza-Cope rearrangement, a testament to the quick facility of the rearrangement.[2]

Due to the mild heating conditions of the reaction carried out, unlike the more stringent ones for a purely hydrocarbon Cope rearrangement, this heteroatomic Cope rearrangement introduced the hypothesis that having a positive charge on a nitrogen in the cope rearrangement significantly reduces the activation barrier for the rearrangement.[2]
Reaction mechanism
Rate acceleration due to positively charged nitrogen
The aza-Cope rearrangements are predicted by the Woodward-Hoffman rules to proceed suprafacially. However, while never explicitly studied, Overman and coworkers have hypothesized that, as with the base-catalyzed oxy-Cope rearrangement, the charged atom distorts the sigmatropic rearrangement from a purely concerted reaction mechanism (as expected in the Cope rearrangement), to one with partial diradical/dipolar character, due to delocalization of the positive charge onto the allylic fragment, which weakens the allylic bond. This results in a lowered activation barrier for bond breaking. Thus the cationic-aza-Cope rearrangement proceeds more quickly than more concerted processes such as the Cope rearrangement.[6][7]
Transition state and stereochemistry
The cationic 2-aza-Cope rearrangement is characterized by its high stereospecificity, which arises from its high preference for a chair transition state. In their exploration of this rearrangement's stereospecificity, Overman and coworkers used logic similar to the classic Doering and Roth experiments,[8] which showed that the Cope rearrangement prefers a chair conformation.[9] By using the cationic 2-aza-Cope/Mannich reaction on pyrrolizidine precursors, they showed that pyrrolizidines with cis substituents from E-alkenes and trans substituents from Z-alkenes are heavily favored, results that are indicative of a chair transition state. If a boat transition state was operative, the opposite results would have been obtained (detailed in image below).[9] As is the trend with many reactions, conversion of the Z-enolate affords lower selectivity due to 1,3 diaxial steric interactions between the enolate and the ring, as well as the fact that substituents prefer quasi-equatorial positioning. This helps explain the higher temperatures required for Z-enolate conversion.[6][9] The boat transition state is even less favored by the cationic-2-aza-Cope rearrangement than it is for the Cope rearrangement: in analogous situations to where the Cope rearrangement takes on a boat transition state, the aza-Cope rearrangement continues in the chair geometry.[1][6][10] These results are in accord with computational chemistry results, which further assert that the transition state is under kinetic control.[11]

Significantly, these stereochemical experiments imply that the cationic 2-aza-Cope rearrangement (as well as Mannich cyclization) occur faster than enol or iminium tautomerization. If they were not, no meaningful stereochemistry would have been observed, highlighting the facility of this fast reaction.[1]
Additional considerations for stereochemistry
The aza-Cope/Mannich reaction, when participating in ring-expanding annulations, follows the stereochemistry dictated by the most favorable chair conformation, which generally places bulky substituents quasi-equatorially. The vinyl and amine components can have either syn or anti relationships when installed on a ring. This relationship is typically dictated by the amine substituent: bulky substituents lead to syn aza-Cope precursors. While anti vinyl and amine substituents generally only have one favored transition state, leading to a cis fused ring system, the favored product of syn substituents can change, dictated by steric interactions with solvents or large N-substituents, which may take preference over bulky substituents and change the transition state.[12][13]
For simple aza-Cope/Mannich reactions that do not participate in ring-expanding annulation, namely condensations of amino alcohols and ethers, bond rotation occurs more quickly than the Mannich cyclization, and racemic products are observed.[14] This can be avoided by using a chiral auxiliary substituent on the amine. Reactions tethered to rings cannot undergo these bond rotations.[1]

Possible thermodynamic sinks for biasing a rearrangement product
Horowitz and Geissman's first example demonstrates a possible thermodynamic sink to couple with the cationic 2-aza-Cope rearrangement, where the product is biased by the phenyl substituent through aryl conjugation, then captured by hydrolysis of the iminium. Other methods of biasing a product include using substituents which are more stable on substituted carbons, releasing ring strain (for instance, by pairing the rearrangement with cyclopropane opening), intramolecular trapping (pictured), and pairing the rearrangement with the Mannich cyclization.[1][15]

The aza-Cope/Mannich reaction
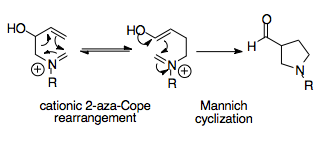
The aza-Cope/Mannich reaction is a synthetically powerful reaction, as it is able to create complex cyclic molecules from simple starting materials. This tandem reaction provides a thermodynamic bias towards one rearrangement product, as the Mannich cyclization is irreversible and its product, an acyl substituted pyrrolidine ring, more stable than that of the rearrangement.[1][16]
The first aza-Cope/Mannich reaction
Overman and coworkers recognized that the cationic 2-aza-Cope rearrangement could potentially be synthetically powerful if an appropriate thermodynamic sink could be introduced. Their logic was to incorporate a nucleophilic substituent into the starting material, namely an alcohol group, which acts only after rearrangement, converted into an enol primed to attack the iminium ion.
This first report of the reaction was a reaction between aldehydes and 2-alkoxy-3-butenamines, which formed an amino alcohol whose aza-Cope/Mannich reaction product was an acyl-substituted pyrrolidine ring. This simple procedure only involved mild heating for several hours. Significantly, the aza-Cope/Mannich reaction occurs in a single step with excellent yield. This procedure is easily applied to condensation of amino ethers (shown below), where the alcohol has been methylated first.[16] After the aza-Cope/Mannich reaction is carried out, the ketone is formed by addition of NaOH.[16] The amine, in this simple case, cannot form the iminium ion from basic ketones; subsequent methods found ways of incorporating ketones into the reaction.[16][17] The utility of this reaction is evident in the fact that even when a less stable isomer is formed, the reaction proceeds, demonstrating its high thermodynamic favorability.[12][17]

Reaction mechanism
The general product of the reaction can potentially occur via two separate pathways: the aza-Cope/Mannich reaction, or an aza-Prins cyclization/pinacol rearrangement. These mechanisms have different stereochemical properties, which elucidate the dominance of the aza-Cope/Mannich reaction. The aza-Cope/Mannich reaction forces each atom in the [1,5] diene analog to undergo sp2 hybridization, erasing the starting material's stereochemistry at the labelled R' position, while the aza-Prins/pinacol rearrangement retains stereochemistry at the labelled R' position, pointing to a simple test that reveals the active mechanism. An enantiomerically pure starting material at the "R'" position should lead to a racemic product if the dominant mechanism is the aza-Cope/Mannich reaction, while the stereochemistry should be retained if the dominant mechanism is an aza-Prins cyclization/pinacol rearrangement pathway. A simple experiment verified that the product was racemic, providing clear evidence of the aza-Cope Mannich reaction as the operative mechanism. Further experiments verified this, using the knowledge that the carbenium ion formed in an aza-Prins/pinacol pathway would be effected by its substituent's ability to stabilize its positive charge, thus changing the reactivity of the pathway. However, a variety of substituents were shown to have little effect on the outcome of the reaction, again pointing to the aza-Cope Mannich reaction as the operative mechanism.[14] Recent literature from the Shanahan lab supports the rare aza-Prins/pinacol pathway only associated with significantly increased alkene nucleophilicity and iminium electrophilicity.[1][6][18][19]
The aza-Cope/Mannich reaction shows high diastereoselectivity, generally in accordance the results of the stereochemical experiments elucidating the transition state of the cationic 2-aza-Cope rearrangement, which follows as this tandem reaction pathway was an integral part of these experiments. The stereochemistry of the rearrangement is slightly more complicated when the allyl and amine substituents are installed on a ring, and thus cis or trans to one another.
The aza-Cope/Mannich reaction starting material, the amino alcohol, can also be thought of as related to the oxy-Cope rearrangement (below), both for its rate acceleration due to ionic involvement, as well as the analogous enol collapsing function of the Mannich cyclization and the keto-enol tautomerization in the oxy-Cope rearrangement.[7]

Synthetic applications of the 2-aza-Cope/Mannich reaction
The aza-Cope/Mannich reaction is often the most efficient way to synthesize pyrrolidine rings, and thus has a number of applications in natural product total syntheses. Because of its diastereoselectivity this reaction has added to the catalog of asymmetric synthesis tools, as seen in the many examples of asymmetric alkaloids synthesized using the reaction. As we have seen in the first aza-Cope/Mannich reaction and in the elucidation of the reaction's stereochemistry, the aza-Cope/Mannich reaction can be used to form pyrrolidine rings and pyrrolizidine rings. It can be used to create many additional ring structures useful in synthesis, such as indolizidine cycles and indole rings.[1][7]
(−)-Strychnine total synthesis
The classic example demonstrating the utility of this reaction is the Overman synthesis of strychnine. Strychnine is a naturally occurring highly poisonous alkaloid, found in the tree and climbing shrub genus Strychnos. Strychnine is commonly used as a small vertebrate pesticide. The first strychnine total synthesis, by R. B. Woodward,[20] represented a major step in natural product synthesis: no molecule approaching its complexity had been synthesized before. The next total syntheses were not reported until the late 1980s, using similar methods, namely by using an intermediate available by degradated strychnine. All of these syntheses used harsh conditions. The Overman synthesis sidesteps these problems, and is the first asymmetric total synthesis of strychnine, taking advantage of the diastereoselectivity and mild reaction conditions of the aza-Cope/Mannich reaction. The aza-Cope/Mannich reaction step proceeded in near quantitative yield. The Overman synthesis is accordingly several orders of magnitude more efficient than its predecessors.[20]

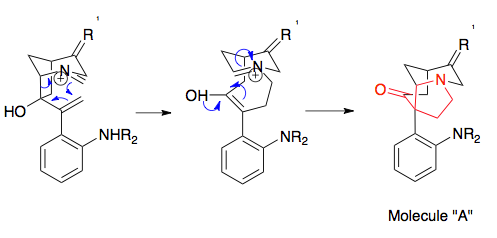
Overman's synthesis of strychnine represents a useful example of the preparation of precursors necessary for the aza-Cope/Mannich rearrangement, representing effective usage of an epoxide ring opening. The key steps of the synthesis of the rearrangement substrate leading to the starting materials necessary for the aza-Cope/Mannich reaction included a Stille reaction to piece together two precursors, an epoxidation of a double bond using tert-Butyl hydroperoxide, a Wittig reaction to convert the ketone to an alkene, and a cyclization step. Amine alkylation (not shown), transforms the molecule to the rearrangement substrate. Significantly, this molecule shows the enantiomeric precision of the aza-Cope/Mannich reaction, as a simple enantiomeric starting material dictates the final enantiomer: the enantiomer of strychnine was produced by using the enantiomer of the starting material.[20][21]

The Overman synthesis, with in-depth details of the synthesis of the rearrangement substrate, as well as the final steps of the reaction, is detailed here: Overman synthesis of (−)-strychnine.
Synthesis of (−)-crinine
Crinine is an alkaloid of the family Amaryllidaceae, and its asymmetric total synthesis was one of the first using the aza-Cope/Mannich reaction. This synthesis represents a significant step in the development of the aza-Cope/Mannich reaction, as it takes advantage of several of the most useful synthetic strategies characteristic of the reaction. This reaction takes advantage of the cationic-2-aza-Cope rearrangement's high diastereoselectivity, as well as usage of the cyanomethyl group to protect the amine during vinyllithium addition and as a leaving group to promote iminium formation, assisted by addition of silver nitrate.[22] This synthesis is one example of many of the cyanomethyl group providing a synthetically useful route towards pyrrolidine and indolizidine formation.

Synthesis of bridged tricyclic alkaloids
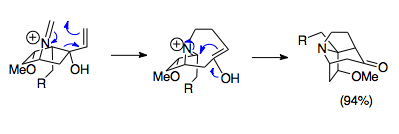
Overman and coworkers developed methods to synthesize complicated bridged tricyclic structures using the aza-Cope/Mannich reaction. These aza-tricyclic structures are found in the complex Stemona alkaloid family, as well as in potential drugs such as some immunosuppressants. The example shown is a facile reaction combining a 1-aza-bicyclo[2.2.1]heptane salt starting material with paraformaldehyde at 80 °C to form the pivotal aza-tricyclic structure of the Stemona alkaloid molecules. Saliently, despite unfavorable orbital overlap due to the sterics of this ring system, the reaction proceeds with 94% yield, highlighting the power of this reaction even under unfavorable conditions.[23]
General ring opening and expansion

The aza-Cope/Mannich reaction, when coupled with existing ring cycles, is often used to create indolizidine cycles (a pyrrolidine connected to a cyclohexane ring). This typical ring annulation, where the cyclopentane moiety is opened with the rearrangement and closed with the Mannich cyclization to form a six membered ring attached to a pyrrolidine ring, while the most popular aza-Cope/Mannich annulation, is not the only one. Seven-membered ring cycles are also possible to synthesize, as the enol and iminium ions stay in close enough proximity to undergo Mannich cyclization.[22] Macrocycle synthesis has not been reported using this reaction, due to the lack of proximity between the enol and iminium.[6] Vinyl oxazolidines can also be used as rearrangement substrates. This rearrangement first creates the vinyl oxazolidine from attack on the cyclohexanone by the aminobutenol, which then undergoes the aza-Cope/Mannich reaction using heat and acid (Lewis or protic). This example breaks and then forms a five-membered ring. More complex examples attach the oxazolidine to another ring, presenting additional methods for the formation of indolizidine cycles.[24]
Scope of the aza-Cope/Mannich reaction
The aza-Cope/Mannich reaction has numerous advantages in comparison to other methods. The gentle conditions of the reaction aren't matched: light heating, normally no higher than 80 °C, a wide range of solvents, and addition of 1 stoichiometric equivalent of acid, commonly camphorsulfonic acid (CSA) or a Lewis acid. Other routes toward pyrrolidine synthesis cannot compete with the stereospecificity, widescale applications in structures containing pyrrolidine derivatives, and large scope of possible starting materials. The reaction exhibits high diastereoselectivity, and is robust, proceeding even when faced with poor orbital overlap in the transition state.[1]

The advantages of the aza-Cope/Mannich reaction have motivated research on the synthesis of the starting materials for the reaction, which split into two main categories: amine addition and iminium formation (red) and installation of the vinyl substituent (blue). A wide variety of N-substituents (R), alkyl and aryl, can be used in the reaction, some of which affect the stereochemical outcome of the reaction. Vinyl groups are generally limited to those which are either 1,1 or 1,2-Disubstituted (vinyl with substituents at R1, and R1,R2 respectively), with a wide range of electronic and steric variety tolerated.[1]
Amine addition and iminium formation
Epoxide ring opening
The ring strain of epoxides provide useful methodology for installation of an amine group two atoms away from an alcohol group. The epoxide may be first broken by bromide nucleophilic attack. Primary amines, aromatic amines, or lithium anilides can also be used as nucleophiles. Protective O-methylation often follows this step and proceeds easily.

When sterics allow for attack on only the appropriate carbon (the terminal carbon as opposed to the second carbon), direct attack by an intramolecular nitrogen is effective, as is the case with strychnine synthesis.[16][25]
Iminium ion formation
The most common way to generate the iminium ion from the installed amine is by adding formaldehyde or paraformaldehyde, which undergoes acid-catalyzed condensation to form the iminium. Overman's strychnine synthesis typifies this method.[6][25] Occasionally, intramolecular carbonyls are used.[9] Other methods for iminium ion formation include using cyanomethyl groups or using oxazolidines as carbonyl precursors.
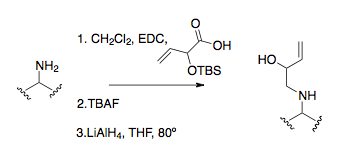
Amine alkylation
Amine alkylation represents a common method to get to imine precursors. Amine alkylation by direct SN2 reaction is only occasionally useful in producing starting materials due to the high propensity of amines to overalkylate.[25] Reductive amination is a more common and effective alkylating procedure, typified in the first aza-Cope rearrangement.[16][26][27] The most useful and standard method of amine alkylation is to have the amine form an amide bond, and subsequently reduce it, often with lithium aluminium hydride.[9]
Oxazolidine use
Ketones and sterically hindered aldehydes are not suitable for the basic aza-Cope/Mannich reaction, as the amine cannot form an iminium ion with them. Dehydrative oxazoline formation followed by heating in the presence of a full equivalent of acid present a way to get around this issue. Overman has reported the use of oxizolidines to generate the iminium ion requisite for the reaction. Upon formation, Overman showed that cyclohexanones can be used for the carbonyl component in pyrrolidine synthesis.[17] This reaction proceeded with various forms of cyclohexanones. When an acyclic ketone was substituted, the reaction proceeded with low yield, highlighting the thermodynamic favorability of releasing cyclohexanone from the double bonded carbonyl, as it creates unfavorable bond strain in the chair conformation. This represents one of the most convenient constructions of the 1-azaspiro[4,5]decane ring system, a useful natural product.[17]
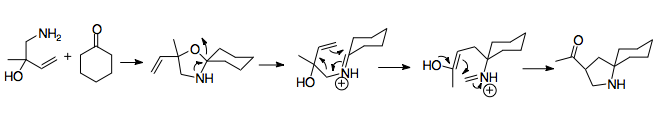
Installation of the vinyl substituent
Vinylation of ketones
Vinylation can offer additional synthetic advantages, allowing for expanded functionality of the reaction.[23] Organolithium reagents are typically used. Often, a substituent or protecting group will be added to the nitrogen, although this isn't always necessary. The addition of lithium to the reaction has a major effect on starting material stereochemistry, as the nitrogen coordinates to it. Starting materials affected by this coordination generally result in anti aza-Cope precursors, while those that aren't, such as those containing highly substituted, sterically hindered amines, result in syn precursors. Thus the nature of the nitrogen substituent is of high importance.[6][25]
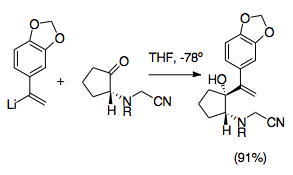
Cyanomethyl group use
Cyanomethyl groups represent an easy way to protect an iminium ion during allylic vinylation of the ketone. Cyanamide groups and analogs have been often used in the generation iminium ions. They are typically installed by nucleophilic addition onto an iminium ion, generally produced by amine alkylation with formaldehyde. The iminium ion is thus masked.[28] It follows that usage of a cyanomethyl group provides an efficient way to control the aza-Cope/Mannich reaction. The cyanomethyl group protects the nitrogen at the 2-position during formation of the other allylic analog by logic similar to cyanide-type umpolung. It then later provides a good leaving group for formation of the iminium ion, in accordance with its usage in iminium ion generation.[29] Iminium ion generation from cyanomethyl groups is normally promoted by addition of silver nitrate, although other silver and copper compounds have been used. This added step allows for more precise control of iminium ion generation.[6][29] Importantly, these preparatory reactions must be carried out at -78 °C to prevent cyanomethyl/vinyllithium interaction. This method also allows for many different possible N-substituents, and can be used to simplify the synthesis of octahydroindoles and pyrroles.[1][29]

The 1- and 3-aza-Cope rearrangements

The 1- and 3-aza-Cope rearrangements are obscure in comparison to the cationic 2-aza-Cope rearrangement due to their activation energies, which are comparatively much higher than that of the cationic 2-aza-Cope rearrangement.
The 1- and 3-aza-Cope have a bias towards imine formation as opposed to enamine formation, as carbon-nitrogen π-bonding is stronger than carbon-nitrogen σ-bonding, meaning the 3-aza-Cope rearrangement is thermodynamically favored, while the 1-aza-Cope rearrangement is not: the imine is nearly 10kcal/mol less in energy. Thus the 3-aza Cope's large activation barriers are kinetically based. Research on both the 1 and 3-aza-Cope rearrangements has focused on finding good driving forces to lowering the activation barriers. Several versions of these rearrangements have been optimized for synthetic utility. The 1-aza-Cope rearrangement is normally paired with thermodynamic driving forces. The 3-aza-Cope rearrangements are generally performed cationically to lower the kinetic barrier to its thermodynamically favorable product.[30]
These rearrangements follow much of the mechanistic logic of the cationic 2-Aza-Cope rearrangement. The 1- and 3-aza-Cope rearrangements both occur preferentially via chair transition states (and retain stereochemistry, similarly to the cationic 2-aza-Cope rearrangement), and are sped up with the introduction of a positive charge, as this gives the transition state more diradical/dipolar character.[30] The 3-aza-Cope rearrangement (and thus also the 1-aza-Cope rearrangement, which goes through the same transition state) is expected to show even less aromatic character in its transition state in comparison to the Cope rearrangement and cationic-2-aza-Cope rearrangement, contributing to the higher temperatures required (close to the temperatures required for the Cope rearrangement, at times even higher, from 170 to 300 degrees) to overcome the kinetic activation barriers for these arrangements.[3][30][31]
The 3-aza-Cope rearrangement

The 3-aza-Cope reaction was discovered soon after the 2-aza-Cope rearrangement was identified, due to its analogous relationship to the Claisen rearrangement. Indeed, in early papers, this version of the aza-Cope rearrangement is often referred to as the amino-Claisen rearrangement, a misrepresentation of the rearrangement, as this would imply that both a nitrogen and oxygen are in the molecule.[3] This rearrangement can be used to form heterocyclic rings involving carbon, most commonly piperidine.
One of the first examples of this arrangement was identified by Burpitt, who recognized the rearrangement occurring in ammonium salts, which, due to their charged nature, proceeded exothermically without addition of heat—importantly, without a tetrasubstituted nitrogen, the rearrangement did not proceed.[32] Following this logic, much of the research on the 3-aza-Cope rearrangement has focused on charged zwitterionic versions of this reaction, as the charge distribution helps lower the activation barrier: in certain cases, the rearrangement can occur at temperatures as low as -20 °C.[33]
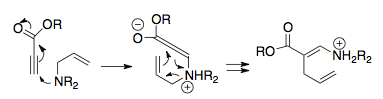
Hill and Gilman first reported a general uncharged 3-aza-Cope rearrangement in 1967. Upon creation of appropriately substituted enamines, intense heating afforded an almost complete rearrangement to the imine product. However, this rearrangement pathway has limited utility.[34]
The 1-aza-Cope rearrangement
The first discovered 1-aza-Cope reaction was a simple analog to the generic Cope reaction and required intense heat to overcome its large thermodynamic activation barrier; most subsequent work on the 1-aza-Cope rearrangement has thus focused on pairing the arrangement with a driving thermodynamic force to avoid these harsh reaction conditions. It has been hypothesized that the 1-aza-Cope rearrangement rate-determining transition state has partial diradical and dipolar transition state character due to the presence of the heteroatom.[4]
Fowler and coworkers have come up with a scheme that mobilizes the 1-aza-Cope rearrangement as a synthetically useful route.[3] Fowler and coworkers recognized that because the barrier for the reaction lies in the nitrogen's thermodynamic preference to stay as an imine, stabilizing the nitrogen could have a thermodynamically beneficial effect. To that end, Fowler and coworkers installed a carbonyl group on the nitrogen, hypothesizing that the lone pair of the nitrogen would be stabilized by participation in an amide bond, and that the electronegativity of this amide group would lower the LUMO of the imine group, making the transition state more favorable.[3] Using this strategy, Fowler and coworkers were able to use the 1-aza-Cope rearrangement to create piperidine and pyridine derivatives. This strategy was shown to be relatively robust, allowing for the formation of products even when forced through a boat transition state, when perturbed with substituent effects, or put in competition with alternative rearrangements.[3] Also significant is the relative ease of production of the reactants, which uses a Diels-Alder reaction paired with relatively simple workup steps, allowing for syntheses using complex cycling.[3]

Other methods of overcoming this thermodynamic barrier include pairing it with cyclopropane ring strain release, which allows the reaction to proceed at much lower temperatures.[35]

References
- ^ a b c d e f g h i j k l m Overman, L. E.; Humphreys, P. G.; Welmaker, G. S. (2011). "The Aza-Cope/Mannich Reaction". Organic Reactions. Vol. 75. pp. 747–820. doi:10.1002/0471264180.or075.04. ISBN 978-0471264187.
- ^ a b c d Horowitz, R. M.; Geissman, T. A. (1950). "A Cleavage Reaction of α-Allylbenzylamines". J. Am. Chem. Soc. 72 (4): 1518–1522. doi:10.1021/ja01160a025.
- ^ a b c d e f g Chu M.; Wu P.L.; Givre S.; Fowler F.W. (1986). "The 1-AZA-Cope rearrangement". Tetrahedron Letters. 27 (4): 461–464. doi:10.1016/S0040-4039(00)85505-7.
- ^ a b Wu, P.L; Fowler, F. W. (1988). "The 1-aza-Cope rearrangement. 2". The Journal of Organic Chemistry. 53 (26): 5998–6005. doi:10.1021/jo00261a003.
- ^ Cook G.R.; Barta N.S.; Stille J.R. (1992). "Lewis acid-promoted 3-aza-Cope rearrangement of N-alkyl-N-allyl enamines". The Journal of Organic Chemistry. 57 (2): 461–467. doi:10.1021/jo00028a016.
- ^ a b c d e f g h i Overman, L.E.; Mendelson, L. T.; Jacobsen, E. J. (1983). "Synthesis applications of aza-Cope rearrangements. 12. Applications of cationic aza-Cope rearrangements for alkaloid synthesis. Stereoselective preparation of cis-3a-aryloctahydroindoles and a new short route to Amaryllidaceae alkaloids". J. Am. Chem. Soc. 105 (22): 6629–6637. doi:10.1021/ja00360a014.
- ^ a b c Overman, L. E. (1992). "Charge as a key component in reaction design. The invention of cationic cyclization reactions of importance in synthesis". Acc. Chem. Res. 25 (8): 352–359. doi:10.1021/ar00020a005.
- ^ Doering, W.v.E.; Roth, W. R. (1962). "The overlap of two allyl radicals or a four-centered transition state in the cope rearrangement". Tetrahedron. 18 (1): 67–74. doi:10.1016/0040-4020(62)80025-8.
- ^ a b c d e Doedens, R. J.; Meier, G.P.; Overman, L.E. (1988). "Synthesis applications of cationic aza-Cope rearrangements. Part 17. Transition-state geometry of [3,3]-sigmatropic rearrangements of iminium ions". J. Org. Chem. 53 (3): 685–690. doi:10.1021/jo00238a039.
- ^ Vogel, E.; Grimme, W.; Dinne, E. (December 1963). "Thermal Equilibrium between cis-1,2-Divinylcyclo-pentane and cis,cis-1,5-Cyclononadiene". Angewandte Chemie International Edition in English. 2 (12): 739–740. doi:10.1002/anie.196307392.
- ^ Lukowski M.; Jacobs K.; Hsueh P.; Lindsay H.A; Milletti M.C. (2009). "Thermodynamic and kinetic factors in the aza-Cope rearrangement of a series of iminium cations". Tetrahedron. 65 (50): 10311–10316. doi:10.1016/j.tet.2009.10.010.
- ^ a b McCann, S. F.; Overman, L. E. (1987). "Medium Effects and the Nature of the Rate-Determining Step in Mannich-Type Cyclizations". J. Am. Chem. Soc. 109 (20): 6107–6114. doi:10.1021/ja00254a033.
- ^ Overman, L. E.; Trenkle, W. C. (1997). "Controlling Stereoselection in Aza-Cope-Mannich Reactions". Isr. J. Chem. 37: 23–30. doi:10.1002/ijch.199700005.
- ^ a b Jacobsen E. J.; Levin J.; Overman L. E. (1988). "Synthesis applications of cationic aza-Cope rearrangements. Part 18. Scope and mechanism of tandem cationic aza-Cope rearrangement-Mannich cyclization reactions". J. Am. Chem. Soc. 110 (13): 4329–4336. doi:10.1021/ja00221a037.
- ^ Marshall, J. A.; Babler, J. H. (1969). "Heterolytic fragmentation of 1-substituted decahydroquinolines". J. Org. Chem. 34 (12): 4186–4188. doi:10.1021/jo01264a104.
- ^ a b c d e f Overman L. E.; Kakimoto, M. (1979). "Carbon-Carbon Bond Formation via Directed 2-Azonia-[3,3]-Sigmatropic Rearrangements. A New Pyrrolidine Synthesis". J. Am. Chem. Soc. 101 (5): 1310–1312. doi:10.1021/ja00499a058.
- ^ a b c d Overman L.E.; Kakimoto M.; Okawara M. (1979). "Directed 2-azonia-[3,3]-sigmatropic rearrangements. a convenient preparation of substituted 1-azaspiro[4,5]decanes". Tetrahedron Letters. 20 (42): 4041–4044. doi:10.1016/s0040-4039(01)86498-4.
- ^ Armstrong, A.; Shanahan, S. E. (2005). "aza-Prins-pinacol approach to 7-azabicyclo[2.2.1]heptanes and ring expansion to [3.2.1]tropanes". Org. Lett. 7: 1335. doi:10.1021/ja00221a037.
- ^ aza-Prins-pinacol approach to 7-azabicyclo[2.2.1]heptanes and ring expansion to [3.2.1]tropanes Armstrong, A.; Shanahan, S. E. Org. Lett. 2005, 7, 1335
- ^ a b c d R. B. Woodward; M. P. Cava; W. D. Ollis; A. Hunger; H. U. Daeniker; K. Schenker (1963). "The total synthesis of strychnine". Tetrahedron. 19 (2): 247–288. doi:10.1016/S0040-4020(01)98529-1. PMID 13305562.
- ^ Knight, S.D.; Overman, L. E.; Pairaudeau, G. (1993). "Synthesis applications of cationic aza-Cope rearrangements. 26. Enantioselective total synthesis of (−)-strychnine". J. Am. Chem. Soc. 115 (20): 9293–9294. doi:10.1021/ja00073a057.
- ^ a b Overman, L. E.; Sugai, s. (1985). "Total Synthesis of (−)-Crinine. Use of Tandem Cationic Aza-Cope Rearrangement/Mannich Cyclizations for the Synthesis of Enantiomerically Pure Amaryllidaceae Alkaloids". Helv. Chim. Acta. 68 (3): 745–749. doi:10.1002/hlca.19850680324.
- ^ a b Brueggemann, M.; McDonald, A. I.; Overman, L.E.; Rosen, M.D.; Schwink, L.; Scott, J.P. (2003). "Total Synthesis of (±)-Didehydrostemofoline (Asparagamine A) and (±)-Isodidehydrostemofoline". J. Am. Chem. Soc. 125 (50): 15284–15285. doi:10.1021/ja0388820. PMID 14664560.
- ^ Overman, L. E.; Shim, J. (1993). "Synthesis applications of cationic aza-Cope rearrangements. Part 25. Total synthesis of Amaryllidaceae alkaloids of the 5,11-methanomorphanthridine type. Efficient total syntheses of (−)-pancracine and (.+-.)-pancracine". Organic Reactions. 58 (17): 4662–4672. doi:10.1021/jo00069a032.
- ^ a b c d Overman L. E.; Kakimoto, M.; Okazaki, M.E.; Meier, G.P. (1983). "Synthesis applications of aza-Cope rearrangements. 11. Carbon-carbon bond formation under mild conditions via tandem cationic aza-Cope rearrangement-Mannich reactions. A convenient synthesis of polysubstituted pyrrolidines". J. Am. Chem. Soc. 105 (22): 6622–6629. doi:10.1021/ja00360a013.
- ^ Overman, L.E.; Fukaya, C. (1980). "Stereoselective total synthesis of (.+-.)-perhydrogephyrotoxin. Synthetic applications of directed 2-azonia-[3,3]-sigmatropic rearrangements". J. Am. Chem. Soc. 102 (4): 1454–1456. doi:10.1021/ja00524a057.
- ^ Borch, R. F.; Bernstein, M. D.; Durst H. D. (1971). "Cyanohydridoborate anion as a selective reducing agent". J. Am. Chem. Soc. 93 (12): 2897–2904. doi:10.1021/ja00741a013.
- ^ Grierson D. S.; Harris, M.; Husson, H.P. (1980). "Synthesis and chemistry of 5,6-dihydropyridinium salt adducts. Synthons for general electrophilic and nucleophilic substitution of the piperidine ring system". J. Am. Chem. Soc. 102 (3): 1064–1082. doi:10.1021/ja00523a026.
- ^ a b c Overman, L. E.; Jacobsen, E. J. (1982). "The cyanomethyl group for nitrogen protection and iminium ion generation in ring-enlarging pyrrolidine annulations. A short synthesis of the amaryllidaceae alkaloid d,1-crinine". Tetrahedron Lett. 67 (51): 2741–2744. doi:10.1016/S0040-4039(00)87446-8.
- ^ a b c Jolidon, S.; Hansen, H. J. (1997). "Untersuchungen über aromatische Amino-Claisen-Umlagerungen". Helv. Chim. Acta. 60 (2): 978–1032. doi:10.1002/hlca.19770600329.
- ^ Zahedi Ehsan; Ali-Asgari Safa; Keley Vahid (2010). "NBO and NICS analysis of the allylic rearrangements (the Cope and 3-aza-Cope rearrangements) of hexa-1,5-diene and N-vinylprop-2-en-1-amine: A DFT study". Central European Journal of Chemistry. 8 (5): 1097–1104. doi:10.2478/s11532-010-0084-1.
- ^ Brannock Kent; Burpitt Robert (1961). "Notes- The Chemistry of Isobutenylamines. II. Alkylation with Allylic and Benzyl Halides". J. Org. Chem. 26 (9): 3576–3577. doi:10.1021/jo01067a645.
- ^ Baxter, E. W.; Labaree, D.; Ammon, H. L.; Mariano, P. S. (1990). "Formal total synthesis of deserpidine demonstrating a versatile amino-Claisen rearrangement/Wenkert cyclization strategy for the preparation of functionalized yohimbane ring systems". J. Am. Chem. Soc. 12 (21): 7682–7692. doi:10.1021/ja00177a032.
- ^ Hill, R. K.; Gilman, N. W. (1967). "A nitrogen analog of the Claisen rearrangement". Tetrahedron Letters. 8 (15): 1421–1423. doi:10.1016/S0040-4039(00)71596-6.
- ^ Boeckman, R. K.; Shair, M.D.; Vargas, R. J.; Stolz, L. A. (1993). "Synthetic and Mechanistic Studies of the retro-Claisen Rearrangement. 2. A Facile route to Medium-Ring Heterocycles via Rearrangement of Vinylcyclopropane- and Cyclobutanecarboxaldehydes". J. Org. Chem. 58 (2): 1295–1297. doi:10.1021/jo00058a001.
Further reading
- Overman, L. E.; Humphreys, P. G.; Welmaker, G. S. (2011). "The Aza-Cope/Mannich Reaction". Organic Reactions. Vol. 75. pp. 747–820. doi:10.1002/0471264180.or075.04. ISBN 978-0471264187.
- Overman, L. E. (2009). "Molecular rearrangements in the construction of complex molecules". Tetrahedron. 65 (33): 6432–6446. doi:10.1016/j.tet.2009.05.067. PMC 2902795. PMID 20640042.
- Siegfried Blechert (1989). "The Hetero-Cope Rearrangement in Organic Synthesis". Synthesis. 1989 (2): 71–82. doi:10.1055/s-1989-27158.