
Morbus Fabry
| Klassifikation nach ICD-10 | |
|---|---|
| E75.2 | Sonstige Sphingolipidosen (inkl. Fabry-(Anderson-)Krankheit) |
| ICD-10 online (WHO-Version 2019) | |

Eine Mutation im GLA-Gen, das für dieses Protein codiert, verursacht bei Morbus-Fabry-Patienten eine verminderte Aktivität dieses Enzyms. Dies hat zur Folge, dass bestimmte Fette (Glycosphingolipide) nicht ausreichend abgebaut werden können und sich in verschiedenen Zellen ansammeln. Diese Ablagerungen führen zu den Symptomen des Morbus Fabry.
Morbus Fabry (auch Fabry-Krankheit, Fabry-Syndrom oder Fabry-Anderson-Krankheit genannt) ist eine seltene, angeborene, monogenetische Stoffwechselstörung aus der Gruppe der lysosomalen Speicherkrankheiten. Bei den betroffenen Patienten fehlt ein Enzym (Katalysator). Morbus Fabry ist erblich und mit Medikamenten behandelbar.
Der Morbus Fabry ist eine Multisystemerkrankung, die eine Vielzahl von Organen des Körpers betreffen kann. Abhängig von den betroffenen Organen können sehr unterschiedliche Symptome auftreten. Die individuell sehr unterschiedliche Ausprägung der Erkrankung und ihre Seltenheit erschweren die Diagnose erheblich, meist wird sie erst viele Jahre nach dem Auftreten der ersten Symptome korrekt gestellt. Die Erkrankung betrifft vor allem das männliche Geschlecht, aber auch heterozygote (mischerbige) Frauen können erkranken. Bei ihnen ist die Erkrankung aber meist weniger stark ausgeprägt und beginnt erst im mittleren Alter klinisch relevant zu werden. Die Lebensqualität der an Morbus Fabry erkrankten Patienten ist häufig deutlich beeinträchtigt.
Mit der Enzymersatztherapie ist die Krankheit seit dem Jahr 2001 kausal behandelbar. Die Patienten erhalten dabei ihr Leben lang gentechnisch produzierte α-Galactosidase A als Infusion. Morbus Fabry ist derzeit nicht heilbar. Unbehandelt erreichen männliche Patienten im Durchschnitt ein Alter von etwa 50, Patientinnen von etwa 70 Jahren. Die Hauptursachen für die frühe Sterblichkeit sind chronisches Nierenversagen, Schädigung des Herzens und eine Beeinträchtigung der Blutversorgung des Gehirns.
Durch eine Mutation (Erbgutveränderung) auf dem X-Chromosom (Geschlechtschromosom) ist die Aktivität des Enzyms α-Galactosidase A stark reduziert. Es ist für den Abbau von zuckerhaltigen Fettstoffen verantwortlich. In den Lysosomen (Recyclingzentren der Zellen) kann vor allem das Stoffwechselprodukt Globotriaosylceramid (auch Gb3 oder auch GL-3 genannt), ein Glycosphingolipid, nicht mehr ausreichend abgebaut werden. Gb3 sammelt sich vor allem in den Zellen der Innenauskleidung der Blutgefäße, den Endothelzellen, an. Im Verlauf der Erkrankung werden diese Ansammlungen pathologisch, das heißt, sie lösen die Fabry-Krankheit aus. Je nach Krankheitsverlauf kann dieser unter Umständen Jahrzehnte dauern.
Die Erkrankung wurde 1898 unabhängig voneinander von dem Deutschen Johannes Fabry und dem Engländer William Anderson erstmals beschrieben.
Epidemiologie
Die Erkrankung betrifft alle Ethnien, und beide Geschlechter können erkranken. Durch ihr seltenes Auftreten ist es schwierig, die Häufigkeit präzise zu bestimmen. In der Literatur werden Prävalenzen von 1 : 476.000[1] über 1 : 117.000[2] bis zu 1 : 40.000[3] genannt. Neuere Studien, die auf den Daten von Neugeborenenscreenings beruhen, deuten allerdings auf eine wesentlich größere Häufigkeit des Morbus Fabry hin.[4] In Oberitalien wurde von 2004 bis 2006 eine Prävalenz von etwa 1 : 3100[5] und in Taiwan eine von etwa 1 : 1500 bei männlichen Neugeborenen[6] ermittelt.[7]
Wird eine Prävalenz von 1 : 3100 zugrunde gelegt, ergeben sich daraus für Deutschland 26.450 Patienten mit Morbus Fabry.[8] Offiziell sind in Deutschland etwa 700 Betroffene bekannt, neben einer wesentlich höheren anzunehmenden Dunkelziffer.[9] Es wird davon ausgegangen, dass bei vielen Patienten die Erkrankung zu Lebzeiten nicht erkannt und das vorzeitige Versterben anderen Erkrankungen zugerechnet wird, so beispielsweise bei isolierten Kardiomyopathien, wie sie bei Morbus-Fabry-Patienten mit einer Restaktivität von α-Galactosidase A auftreten können.[10] Ein weiteres Indiz dafür ist, dass in Taiwan bei 86 % der positiv getesteten Neugeborenen eine kryptische Spleißmutation vom Typ IVS4+919G > A besteht, die zuvor vor allem bei Morbus-Fabry-Patienten mit dem kardialen Phänotyp gefunden wurde.[6] Bei diesen Patienten äußert sich der Morbus Fabry im Wesentlichen in einer Herzmuskelerkrankung (Kardiomyopathie). Die intronische Form dieser Mutation liegt bei vielen taiwanischen Patienten mit hypertropher Kardiomyopathie vor.[7][11]
In bestimmten Subpopulationen, bei denen es einen Zusammenhang mit Symptomen von Morbus Fabry gibt, ist die Inzidenz naturgemäß höher. In einer Studie mit 911 spanischen Hämodialyse-Patienten wurden vier männliche und drei weibliche mit Veränderungen im GLA-Gen ermittelt, was einer Prävalenz von 1 : 182 bei dieser Subpopulation entspricht.[12]
Genetik und Molekularbiologie


A Ein 17-jähriges Mädchen, das eine Transversion von Thymin nach Guanin in Exon 6, Position 884 aufweist. Diese Nukleotidsubstitution ändert das Codon TTC, das für die Aminosäure Phenylalanin codiert, in TGC, wodurch bei der Translation Cystein in das Genprodukt α-Galactosidase A eingebaut wird. Folglich findet sich in der α-Galactosidase A dieser Patientin in Position 295 Cystein statt Phenylalanin. Es liegt eine p.Phe295Cys-Mutation vor.
B Eine 46-jährige Frau, in deren GLA-Gen in Exon 1, Position 125, ebenfalls eine Transversion von T nach G vorliegt. Das ATG-Codon, das für Methionin codiert, wird daher zu AGG, was für die Aminosäure Arginin steht, die sich dann nach der Translation in Position 42 der α-Galactosidase A wiederfindet. Es ist eine p.Met42Arg-Mutation.
C Bei dieser 63-jährigen Patientin findet sich in Exon 6, Position 982, eine G-T-Transversion. Durch den Nukleotid-Austausch wird das GGG-Codon zu TGG und aus Glycin wird Tryptophan, das sich dann auf Position 328 der α-Galactosidase A findet. Die Mutation wird folglich mit p.Gly328Trp bezeichnet.

Morbus Fabry ist eine Erkrankung, die auf einem Gendefekt (Mutation) des weiblichen Geschlechtschromosoms, des X-Chromosoms, beruht.[14] Jeder von der Erkrankung betroffene Vater vererbt die Erkrankung an alle seine Töchter, während alle seine Söhne gesund bleiben. Trägt die Mutter das mutierte Gen, so haben ihre Kinder – unabhängig vom Geschlecht – ein 50%iges Risiko, die Erkrankung zu erben. Von der Mutation ist das GLA-Gen betroffen, das sich auf dem langen Arm des X-Chromosoms am Genlocus q22.1 befindet.[15]
Das Genprodukt ist ein Homodimer und wird wie alle lysosomalen Enzyme cotranslational, das heißt während der Übersetzung der mRNA in die Aminosäuresequenz, mit einem Mannose-6-phosphat-Rest versehen. Ein Teil der phosphorylierten α-Galactosidase-A-Moleküle wird von den Zellen sezerniert und von anderen Zellen über den membranständigen Mannose-6-phosphat-Rezeptor per Endozytose aufgenommen. Die Wiederaufnahme der phosphorylierten α-Galactosidase A über den Mannose-6-phosphat-Rezeptor ist die Grundlage für die Enzymersatztherapie.[16]
Besonderheiten des X-chromosomalen Erbgangs
Bedingt durch den X-chromosomalen Erbgang ist die Krankheit bei Männern und Frauen unterschiedlich stark ausgeprägt. Männliche Patienten werden als hemizygote und weibliche als heterozygote Merkmalsträger bezeichnet.[17] Früher ging man davon aus, dass nur Männer an Morbus Fabry erkranken könnten und dass heterozygote Frauen lediglich Überträgerinnen seien. Dies ist bei der bei weitem überwiegenden Mehrzahl anderer X-chromosomaler Erbkrankheiten – wie beispielsweise bei der Bluterkrankheit oder der Muskeldystrophie vom Typ Duchenne – der Fall.[18][19] Mittlerweile weiß man, dass auch Frauen, die dieses Merkmal heterozygot haben, am Morbus Fabry erkranken können. Einige Autoren empfehlen daher, von der Bezeichnung X-chromosomal-rezessiv abzusehen, da diese irreführend ist. Stattdessen wird die Terminologie X-chromosomale Vererbung empfohlen (engl. X-linked inheritance).[19][20][21][22]
Bei heterozygoten Patientinnen sind in jeder kernhaltigen Körperzelle mit DNA ein nicht mutiertes und ein mutiertes X-Chromosom vorhanden. Durch die X-Inaktivierung wird in jeder Zelle eines der beiden X-Chromosomen inaktiviert. Diese Inaktivierung erfolgt in jeder Zelle eigenständig und nach dem Zufallsprinzip (so genanntes Mosaik). Rein statistisch werden daher 50 % der Zellen α-Galactosidase A ohne oder mit verminderter Aktivität produzieren – je nach Art der Mutation. Die anderen 50 % der Zellen produzieren α-Galactosidase A mit einer normalen Aktivität („gesunde Zellen“). Ein Teil der aktiven α-Galactosidase A wird von den „mutierten Zellen“ mit dem aktivierten X-Chromosom, auf dem das defekte GLA-Gen sitzt, wie zuvor beschrieben per Endozytose aufgenommen. Dieser Enzymtransfer ist zwar ausreichend, um eine Eliminierung der mutierten Zellen durch das Immunsystem zu verhindern, aber zu gering, um den Gendefekt zu kompensieren, damit die Akkumulation von Globotriaosylceramiden verhindert wird.[23] Im Vergleich zu anderen lysosomalen Enzymen ist die Aufnahme von α-Galactosidase A mittels Enzymtransfer relativ niedrig.[24]
Mit der X-Inaktivierung lässt sich zwar erklären, dass im Mittel bei heterozygoten Frauen die Erkrankung deutlich später symptomatisch wird und weniger stark als bei Männern ausgeprägt ist. Sie ist aber kein ausreichendes Modell, um die große Bandbreite an unterschiedlichen Ausprägungen der Krankheit bei Frauen zu verstehen. Beispielsweise benötigen etwa 10 % der Patientinnen im Laufe der Progression der Krankheit eine Nierenersatztherapie, was dem „klassischen Phänotyp“ bei Männern entspricht. Andere heterozygote Frauen bleiben dagegen weitgehend symptomfrei. Die Ursache hierfür ist noch nicht vollständig geklärt.[7]
Eine Hypothese geht davon aus, dass Unregelmäßigkeiten bei der Inaktivierung des X-Chromosoms eine wichtige Rolle für die Variationsbreite beim heterozygoten Phänotyp spielen. Man spricht dabei von einer „schiefen (engl. skewed) X-Inaktivierung“, bei der das statistisch zu erwartende 50:50-Verhältnis zwischen „mutierten“ und „gesunden“ Zellen deutlich verschoben ist. Diese Verschiebung wird nicht durch einen Wachstumsvorteil der mutierten Zellen hervorgerufen.[25] Ein Indiz für die schiefe X-Inaktivierung sind heterozygote Patientinnen mit einem Phänotyp, bei dem der Morbus Fabry voll entwickelt ist, da bei über 95 % der Zellen das mutierte X-Chromosom aktiviert ist. Etwa eine von 200 Morbus-Fabry-Patientinnen entspricht diesem Phänotyp. Ein weiteres Indiz für die schiefe X-Inaktivierung ist ein weibliches heterozygotes eineiiges Zwillingspaar, bei dem einer der Zwillinge symptomfrei und der andere klinisch relevant ist.[26] In einer Studie mit 28 Patientinnen konnte zwar bei den meisten Patientinnen eine schiefe X-Inaktivierung in den Leukozyten festgestellt werden, sie stand jedoch in keiner Relation zur klinischen Manifestation der Krankheit oder zur verbliebenen Restenzymaktivität. Die Autoren sehen deshalb keinen Zusammenhang zwischen Phänotyp und schiefer X-Aktivierung.[27]
Mutationsvarianten
Die durch Mutationen hervorgerufenen Defekte im GLA-Gen sind sehr heterogen. Bisher wurden über 500 verschiedene Mutationen erfasst. Darunter sind Punktmutationen vom Typ missense und nonsense, Spleißmutationen, kleine Deletionen und Insertionen sowie große Deletionen.[7] Am häufigsten sind Punktmutationen (ca. 71 %), gefolgt von kleinen Deletionen und Insertionen, die weniger als 60 Nukleotide betreffen (ca. 27 %) und großen Deletionen, die ein oder mehrere Exons betreffen (ca. 2 %).[28] In den meisten Fällen führt die Mutation zu einem vollständigen Verlust der Enzymaktivität.[29] Einige Mutationen, die zu Veränderungen in der α-Galactosidase A führen und einen ausreichend großen Abstand vom aktiven Bereich des Enzyms haben, führen nur zu kleinen strukturellen Veränderungen des Enzyms, so dass eine bestimmte Restaktivität des Enzyms noch vorhanden ist. Solche Mutationen, wie beispielsweise p.Met72Val, p.Gln279Glu oder p.Met296Ile, sind durch einen milden Krankheitsphänotyp gekennzeichnet. Die Genprodukte weisen zwar normale Werte für die Michaeliskonstante Km und die Umsatzgeschwindigkeit Vmax auf, allerdings werden diese mutierten Enzyme posttranslational deaktiviert und anschließend schnell abgebaut. Durch Galactose kann die Stabilität dieser mutierten Enzyme in Lymphozyten offensichtlich erhöht werden.[28]
Einen ausgesprochenen Hotspot, das ist ein für Mutationen besonders anfälliger Bereich, gibt es auf dem GLA-Gen nicht. Auffällig sind gehäufte DNA-Umgruppierungen (DNA Rearrangements) in Exon 7, das offensichtlich eine erhöhte Anfälligkeit für Umgruppierungen aufweist.[28]
Pathologie



Der Morbus Fabry gehört zu der mindestens 50 Mitglieder umfassenden Gruppe der lysosomalen Speicherkrankheiten und dort zur Untergruppe der Sphingolipidosen. Die Erkrankung beruht auf einem Mangel des lysosomalen Enzyms α-Galactosidase A. Durch dieses Defizit sammeln sich bestimmte Stoffwechselprodukte, wie beispielsweise Globotriaosylceramid (Gb3, Gl3, früher auch Ceramid-trihexosid genannt), in den Endothelzellen verschiedener Organsysteme an. Die verminderte Aktivität der α-Galactosidase führt im Wesentlichen zu einer Anreicherung von Globotriaosylceramid. Daneben reichert sich auch Digalactosaylceramid – vor allem in den Nieren – und Globotriaosylshingosin (lyso-Gb3, lyso-Gl3) an.[30] Diese Sphingolipide sind wichtige Bestandteile der Zellmembran.
Die genauen Zusammenhänge zwischen verminderter oder gar völlig fehlender Aktivität der α-Galactosidase A und den pathologischen Vorgängen in den betroffenen Organen – die letztlich zur Fabry-Krankheit führen – sind noch unzureichend aufgeklärt.[31] Die Vielfältigkeit der betroffenen Organe lässt darauf schließen, dass sekundäre biochemische Mechanismen, in der Sphingolipide eine Rolle spielen, den Verlauf der Erkrankung bestimmen.[32][33]
Die im nachfolgenden Kapitel beschriebenen Symptome wie beispielsweise progredientes chronisches Nierenversagen werden in vielen Publikationen der Akkumulation von Globotriaosylceramid im Lysosom von Endothelzellen zugeschrieben.[3] Eine Reihe von klinischen Effekten, insbesondere bei der Enzymersatztherapie des Morbus Fabry, passen jedoch nicht zu dieser offensichtlich vereinfachten Modellvorstellung. So sind beispielsweise bei einem Teil der Patienten progressive Komplikationen zu beobachten,[34] was darauf schließen lässt, dass es keine direkte Korrelation zwischen Gb3 und klinischer Manifestation des Morbus Fabry gibt. Zu dem vereinfachten Modell passt auch nicht die Beobachtung, dass ein großer Teil weiblicher GLA-Mutationsträger Symptome entwickelt, die denen hemizygoter Patienten ähneln, obwohl diese Patientinnen beachtliche Mengen an zirkulierendem Enzym aufweisen.[35][36][37][38][39] Zudem beginnt die Ansammlung von Gb3 im Lysosom bei hemizygoten Patienten schon im frühesten Kindesalter beziehungsweise vorgeburtlich, lange bevor sich klinisch relevante Symptome entwickeln.[40] Es gibt auch weder bei hemizygoten noch bei heterozygoten Patienten eine Korrelation zwischen dem Erkrankungsgrad und dem Plasma- oder Urinspiegel von Gb3.[39][41][42]
Da selbst bei Patienten ohne jegliche Aktivität der α-Galactosidase A sich die Krankheit nicht im Kindesalter manifestiert, geht man davon aus, dass die Akkumulation von Gb3 nicht die unmittelbare Ursache für den Morbus Fabry ist.[43] Aktuell nimmt man an, dass Globotriaosylsphingosin – ein Stoffwechselprodukt von Globotriaosylceramid – letztlich die Ursache für die pathologischen Schäden beim Morbus Fabry ist. Zumindest bei der Schädigung der Glomeruli, die zur Niereninsuffizienz beim Morbus Fabry führt, spielt lyso-Gb3 eine entscheidende Rolle. Lyso-Gb3 setzt TGF-β1 und den Makrophagen-Inhibitor CD74 frei. Der darauf folgende Pathomechanismus gleicht dem einer diabetischen Nephropathie.[7][31]
Klinisches Bild


Verlaufsformen und Schweregrad
Bei den Patienten und den Verlaufsformen des Morbus Fabry unterscheidet man zwei Arten: die „klassischen“ hemizygoten Patienten, bei denen die α-Galactosidase A keinerlei Aktivität hat, und die „atypischen“ heterozygoten Patientinnen, bei denen noch eine Restaktivität des Enzyms vorhanden ist. Der klassische Krankheitsverlauf äußert sich in einem frühen Auftreten der Symptome, meist in mehreren Organen. Deutlich später treten dagegen die Symptome bei den atypischen heterozygoten Patienten auf. Zudem kann in solchen Fällen die Krankheit lokal, beispielsweise auf den Herzmuskel, begrenzt sein.[14] Männliche Patienten entwickeln schon ab dem Kindesalter die für Morbus Fabry typischen Symptome. Bei Frauen ist dies häufig erst in einem Alter von 40 bis 50 Jahren der Fall. Durch die Restaktivität an α-Galactosidase A sind die Symptome häufig auch weniger stark ausgeprägt.
Die Symptome sind vielschichtig und können individuell unterschiedlich auftreten. Für die Diagnosestellung sind die frühen Symptome von großer Bedeutung. Die meisten späten Symptome bestimmen dagegen die Mortalität (Sterberate) der Patienten.
Der Severity Index ist eine Kennzahl, die den Schweregrad einer Erkrankung bestimmt. Für männliche Morbus-Fabry-Patienten wurde die Penetranz mit 100 % und der Schweregrad mit 84 % eingestuft. Für Patientinnen liegen die Werte für die Penetranz bei 70 % und für den Schweregrad bei 4 %.[21][19]
Lebensqualität
Die Symptome der Erkrankung bewirken vor allem bei den männlichen Patienten eine Lebensqualität auf einem niedrigen Niveau.[44] Es ist vergleichbar mit dem von AIDS-Patienten.[10] Bei Morbus-Fabry-Patientinnen ist die Lebensqualität auf einem ähnlichen Niveau wie bei Patienten mit Multipler Sklerose oder rheumatoider Arthritis.[45]
Der Morbus Fabry hat erhebliche negative Einflüsse auf das psychosoziale Umfeld der Betroffenen. Über die Hälfte der männlichen Patienten ist unverheiratet. Ein hoher Anteil ist ohne Arbeitsplatz.[46] Depressionen sind bei Morbus-Fabry-Patienten ausgesprochen häufig. Sie werden unterdiagnostiziert beziehungsweise unterbehandelt und mindern die Lebensqualität der Patienten signifikant. Einer Studie zufolge leiden 46 % der Patienten unter Depressionen und bei 28 % sind diese so schwerwiegend, dass sie klinisch relevant sind. Auf der Hamilton-Skala werden dabei Werte über 26 erreicht.[47] Im Gegensatz zur Normalbevölkerung ist der Anteil an Männern mit schweren Depressionen (36 %) höher als bei Frauen (22 %).[48]
Mehrere Studien empfehlen die psychiatrische und neuropsychologische Untersuchung von Patienten mit Morbus Fabry.[7][45][49] Im Detail beklagen die Patienten körperliche Beschwerden, Traurigkeit und seelisches Leid. Unter Stress nehmen die körperlichen Beschwerden zu. Psychologische Tests zeigen überdurchschnittliche Verhaltensstörungen, Misstrauen, Abwehrhaltungen, emotionale Aufwühlung und das Gefühl der Isoliertheit. Die Ergebnisse dieser Tests gleichen weitgehend denen von Schmerzpatienten.[50]
Frühe Zeichen und Symptome


Schmerzen
Eines der ersten Symptome bei der klassischen Verlaufsform des Morbus Fabry sind Schmerzen in Händen und Füßen, den Akren. Diese Akroparästhesien treten bereits in der Kindheit auf. Sie werden durch Schäden an den dünnen Nervenfasern (Small-Fiber-Neuropathie) des vegetativen[51] und des peripheren somatischen Nervensystems[52] hervorgerufen. Etwa 60 bis 80 % der Jungen und Mädchen mit der klassischen Verlaufsform sind von diesen Schmerzen betroffen.[53][54]
Zwei Arten von Schmerzen werden von den Patienten beschrieben: periodisch wiederkehrende anfallartige Schmerzattacken, auch „Fabry-Krisen“ genannt, mit brennenden Schmerzen, die von den Händen und Füßen ausgehend in andere Körperteile ausstrahlen, und chronische Schmerzen, die brennenden und kribbelnden Parästhesien entsprechen.[55] Die Fabry-Krisen können durch Fieber, sportliche Betätigung, Stress, Erschöpfung und schnelle Temperaturwechsel ausgelöst werden.[56] Diese Symptome werden teils als rheumatische Beschwerden, Raynaud-Syndrom, systemischer Lupus erythematodes und vor allem als Wachstumsschmerzen fehlinterpretiert.
Die Schmerzen lassen im Erwachsenenalter meist nach. Sie treten bei Jungen früher und häufiger als bei Mädchen auf. Bei Jungen durchschnittlich im siebten Lebensjahr, bei Mädchen im neunten.[54] Die Schmerzen haben einen erheblichen negativen Einfluss auf die Lebensqualität der Patienten.[7][44][48]
Gastrointestinale Beschwerden
Beschwerden des Verdauungstraktes sind ein weiteres häufiges, meist unterschätztes,[57] frühes Symptom des Morbus Fabry. Diese Leiden bleiben meist auch noch im Erwachsenenalter erhalten. Die Patienten beklagen Bauchschmerzen, meist nach dem Essen, Durchfall, Übelkeit und Erbrechen. Dies kann wiederum die Ursache für Appetitlosigkeit sein. Verursacht werden diese gastrointestinalen Symptome vermutlich von Gb3-Ablagerungen in den autonomen Ganglien (Ganglia autonomica) des Darms und den mesenterischen Blutgefäßen.[58]
Anhidrose
Viele Morbus-Fabry-Patienten können keinen Schweiß absondern (Anhidrose)[59] oder dies nur stark vermindert tun (Hypohidrose).[60] Die Werte für die Hautimpedanz sind daher vergleichsweise hoch.[61] Die Anhidrose/Hypohidrose kann bei den Betroffenen zu einer Wärmeintoleranz[62] und erheblichen Einschränkungen bei sportlichen Betätigungen führen.[7][63] In einer Studie mit 714 Morbus-Fabry-Patienten wurde bei 53 % der männlichen und bei 28 % der weiblichen Probanden eine Anhidrose diagnostiziert.[60] Die Ursache für die verminderte Fähigkeit der Schweißabsonderung sind die Lipid-Ansammlungen innerhalb der Neuronen des vegetativen Nervensystems.[61]
Angiokeratome
Das am leichtesten zu erkennende Frühsymptom bei Morbus Fabry sind Angiokeratome. Dies sind gutartige rot-violette Hautveränderungen mit leichten Erhebungen. Sie werden typischerweise an Gesäß, Leistenregion, Bauchnabel und Oberschenkel gebildet. Gelegentlich sind auch die Schleimhäute, beispielsweise im Mund, betroffen. In den meisten Fällen handelt es sich bei den Angiokeratomen um kleine oberflächliche Angiome, die durch Schäden des vaskulären Endothels der Haut, verbunden mit Vasodilatationen in der Haut, entstanden sind. Sie nehmen in Anzahl und Größe mit dem Alter zu und können einzeln oder in Gruppen auftreten.[60][64] Neben den Angiomen wurden auch Fälle mit Teleangiektasien[60][62] und subkutanen Ödemen[65] als Ursache der Angiokeratome berichtet.[7]
 Histologisches Präparat einer mittels Biopsie entnommenen Hautprobe eines Patienten mit Morbus Fabry.
Histologisches Präparat einer mittels Biopsie entnommenen Hautprobe eines Patienten mit Morbus Fabry.
In der lichtmikroskopischen Aufnahme lassen sich die typischen Hautläsionen als kleine oberflächliche Angiome erkennen.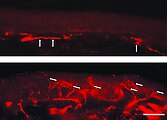 Fluoreszenzmikroskopaufnahmen eines Gefrierschnittes der Haut eines Morbus-Fabry-Patienten.
Fluoreszenzmikroskopaufnahmen eines Gefrierschnittes der Haut eines Morbus-Fabry-Patienten.
Auffällig ist der Mangel an intraepidermalen Nervenfasern und das Vorhandensein von Fasern, die zum subepidermalen Nervenplexus gehören (Pfeile). Die untere Hautprobe stammt dagegen vom Rücken des Patienten. Hier ist die dichte Innervation der Epidermis (Pfeile) auffällig. Zeichnung nach einer Mikroskopbetrachtung einer Carminfärbung der Epidermis mit einem Angiokeratom, Aquarell aus Johannes Fabrys Artikel von 1898
Zeichnung nach einer Mikroskopbetrachtung einer Carminfärbung der Epidermis mit einem Angiokeratom, Aquarell aus Johannes Fabrys Artikel von 1898



Vortexkeratopathie

Charakteristische Hornhauttrübungen sind das häufigste frühe Symptom des Morbus Fabry. Sie können an der Spaltlampe sicher diagnostiziert werden und treten bei nahezu allen hemizygoten Patienten auf.[66][67][68] Diese Form der Hornhauttrübung wird als Cornea verticillata oder Vortexkeratopathie bezeichnet. Sie tritt beidseitig auf und hat ein charakteristisches cremefarbenes wirbelartiges Muster. Die Trübung beeinträchtigt die Sehschärfe nicht. Einige Arzneimittel wie Amiodaron und Chloroquin erzeugen bei längerer Einnahme ebenfalls Vortexkeratopathien.[7][69]
Späte Symptome
Nierenschädigung

Wie die meisten Symptome bei Morbus Fabry weist auch die Schädigung der Nieren einen progressiven Verlauf auf. Er endet mit dem terminalen Nierenversagen und bewirkt eine deutlich reduzierte Lebenserwartung.[70] Bei dem klassischen Krankheitsbild des Morbus Fabry führen die Gb3-Ansammlungen in den Endothelzellen des Glomerulums, in den Mesangiumzellen, in den Podozyten und in den Zellen des Interstitiums zur Schädigung der Niere.[71] Bei diesen Zellen handelt es sich um ausdifferenzierte Epithelzellen. Auch im Epithel der Henle-Schleife und des distalen Tubulus sowie im Endothel und den Zellen der glatten Muskulatur der Arteriolen der Niere finden sich Glycosphingolipid-Ansammlungen.[72][73] Im Transmissionselektronenmikroskop (TEM) sind die Gb3-Ablagerungen im Zytoplasma gut zu erkennen. Sie haben die Form von Myelinstrukturen und stoßen an den Zellkern. Mit zunehmender Gb3-Akkumulation wird das Mesangium aufgeweitet, worauf eine segmentale oder globale Glomerulosklerose mit Eindickung der Basalmembranen erfolgt.[74] Mikrovaskuläre Läsionen und Schädigungen der für die Filterleistung wichtigen Podozyten sowie der Epithelzellen des Tubulus werden dabei als mögliche Mechanismen diskutiert.[75]
Die Nierenschädigung beginnt beim klassischen Krankheitsverlauf in der zweiten bis dritten Lebensdekade. Zunächst ist eine Mikroalbuminurie, das ist die Ausscheidung geringer Mengen des Proteins Albumin über den Urin, zu beobachten, die sich zu einer Proteinurie (die Ausscheidung größerer Mengen an Eiweiß über den Urin) weiterentwickelt. Der Verlauf ähnelt einer diabetischen Nephropathie und trägt unmittelbar zur Progression der Fabry-Nephropathie bei. Mit zunehmendem Alter wird die Proteinurie immer schwerwiegender.[72] Im weiteren Verlauf entwickelt sich eine Isosthenurie, das heißt, die Nieren verlieren ihre Konzentrations- oder Verdünnungsfähigkeit vollständig. Damit einher gehen Veränderungen bei der tubulären Rückresorption, Sekretion und Exkretion.
Anfänglich wird die Nierenschädigung durch eine glomeruläre Hyperfiltration überdeckt. Ist aber erst einmal eine kritische Anzahl von Nephronen geschädigt, so nimmt die Nierenfunktion progressiv ab. Die glomeruläre Filtrationsrate, ein Maß für die Filtrationsleistung der Nieren, nimmt unbehandelt pro Jahr um etwa 12 ml/min ab.[75] In der dritten bis fünften Lebensdekade verschlechtert sich die Nierenfunktion allmählich und eine renale Azotämie – dies ist die abnorme Vermehrung von stickstoffhaltigen Stoffwechselprodukten wie beispielsweise Harnstoff und Kreatinin im Blut – tritt ein.[76] In diesem Stadium dominieren Fibrose, Sklerose und tubuläre Atrophie die Fabry-Nephropathie und führen letztlich zum terminalen Nierenversagen, das bei männlichen Patienten in der vierten bis fünften Dekade eintritt.[77][78] Etwa 17 % aller männlichen und 1 % aller weiblichen Morbus-Fabry-Patienten entwickeln eine terminale Niereninsuffizienz und werden dialysepflichtig.[75] Dabei ist die Hälfte der Patienten unter 53 Jahre alt.[77] Mehr als die Hälfte der Morbus-Fabry-Patienten entwickelt im Krankheitsverlauf eine Nephropathie.[79] Das terminale Nierenversagen ist ein wesentlicher Faktor für die Morbidität und Mortalität. Ohne Nierenersatztherapie führt eine Urämie (Harnvergiftung) unweigerlich zum Tod.[7][78]
- Gewebeproben aus der Niere von Patienten mit Morbus Fabry
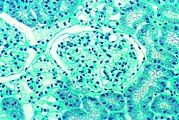 Diese lichtmikroskopische Aufnahme zeigt die Akkumulation von Gb3 in den Endothelien des Glomerulums, in den Mesangiumzellen, den Zellen im Interstitium und in den Podozyten.
Diese lichtmikroskopische Aufnahme zeigt die Akkumulation von Gb3 in den Endothelien des Glomerulums, in den Mesangiumzellen, den Zellen im Interstitium und in den Podozyten.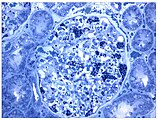 Ebenfalls eine lichtmikroskopische Aufnahme. In den Podozyten ist die erhöhte Ansammlung an Gb3 durch eine violette Anfärbung sichtbar gemacht worden.
Ebenfalls eine lichtmikroskopische Aufnahme. In den Podozyten ist die erhöhte Ansammlung an Gb3 durch eine violette Anfärbung sichtbar gemacht worden. Die TEM-Aufnahme zeigt die massive elektronendichte (= schwarze) Ansammlung an Glycosphingolipiden im Lysosom der Podozyten.
Die TEM-Aufnahme zeigt die massive elektronendichte (= schwarze) Ansammlung an Glycosphingolipiden im Lysosom der Podozyten. Ebenfalls eine TEM-Aufnahme. Sie zeigt die Inklusionen von Glycosphingolipiden unterschiedlicher Form und Größe in den Zellen des distalen Tubulus.
Ebenfalls eine TEM-Aufnahme. Sie zeigt die Inklusionen von Glycosphingolipiden unterschiedlicher Form und Größe in den Zellen des distalen Tubulus.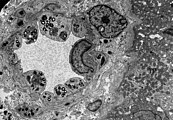 TEM-Aufnahme von den Endothelien und Zellen der glatten Muskulatur einer Nierenarteriole mit Inklusionen von Glycosphingolipiden
TEM-Aufnahme von den Endothelien und Zellen der glatten Muskulatur einer Nierenarteriole mit Inklusionen von Glycosphingolipiden





Herzschädigung

A Linksventrikuläre Hypertrophie (LVH) bei einem 51-jährigen dialysepflichtigen Patienten mit zerebrovaskulärer Beteiligung.
B 56-jähriger Patient mit hypertropher Kardiomyopathie und Herzrhythmusstörung, Leukoaraiose sowie Transplantatniere.
C Late-Enhancement-Messung nach der Gabe eines gadoliniumhaltigen Kontrastmittels bei einer 63-jährigen dialysepflichtigen Patientin.
Etwa 40 bis 60 % der Morbus-Fabry-Patienten zeigen kardiale Symptome wie linksventrikuläre Hypertrophie (LVH, Verdickung der Herzwände der linken Herzkammer), Herzrhythmusstörung (Arrhythmie), Angina Pectoris (anfallsartiger Schmerz in der Brust) und Dyspnoe (erschwerte Atemtätigkeit).[78][80][81][82][83] Die Herzrhythmusstörung und die beeinträchtigte Herzfrequenzvariabilität werden vom Sinusknoten, dem Erregungsleitungssystem, und einer Störung des Gleichgewichts zwischen sympathischem und parasympathischem Tonus hervorgerufen. Diastolische Dysfunktionen und linksventrikuläre Hypertrophien sind wichtige Symptome des Morbus Fabry. Für Männer sind diese Symptome grundsätzlich schwerwiegender als für Frauen. Myokardischämien (Durchblutungsstörungen des Herzmuskels) sind die Folge einer schlechten Koronardurchblutung.[84]
Im Alter entwickeln sich progredient Myokardfibrosen, die sowohl reversibel interstitiell als auch irreversibel narbig (replacement fibrosis) sind.[85][86] Die irreversiblen narbigen Fibrosen bilden sich in fast allen Fällen zuerst in der hinteren seitlichen Herzwand und im Midmyokard. Bei Patienten im Endstadium reduziert eine transmurale (die gesamte Dicke der Wandschicht des Herzens betreffende) narbige Fibrose allmählich die Herzfunktion bis zur kongestiven Herzinsuffizienz.[87][88][89][90] Maligne Arrhythmien sind für die meisten Fälle von Herztod bei Morbus-Fabry-Patienten verantwortlich.[7][83][90]
Die linksventrikulären strukturellen Veränderungen des Herzens finden sich bei Morbus-Fabry-Patienten häufig. Mittels Echokardiografie (Ultraschalluntersuchung des Herzens) oder kardialer Magnetresonanztomographie (MRT) können die meist konzentrischen Hypertrophien[80][81] sichtbar gemacht werden. Da mit zunehmendem Alter durch die Ersatzfibrose die linksventrikuläre Hinterwand des Herzens immer dünner wird, ist die Messung der Septumdicke – das ist die Stärke der Scheidewand zwischen linker und rechter Herzhälfte – besonders wichtig. Unabhängig von den strukturellen Veränderungen scheint die Systole, die Phase, in der das Blut aus der linken und rechten Herzkammer herausgepresst wird, bei der Messung mit konventionellen Methoden weitgehend erhalten zu bleiben.[80][81][82][91] Die durch Morbus Fabry verursachte Kardiomyopathie ist durch eine reduzierte Kontraktion und Relaxation des Herzmuskels gekennzeichnet. Gewebedoppler (sowohl tissue velocity imaging als auch strain rate imaging) kann die Herzmuskelfunktion quantifizieren.[7][92] Mit dieser Methode kann die Kardiomyopathie noch vor der Entwicklung einer linksventrikulären Hypertrophie diagnostiziert werden.[91][93]
- Echokardiografien von Patienten mit Morbus Fabry
 Parasternale lange Achse: Deutlich sichtbar die linksventrikuläre Hypertrophie mit erhöhter Septumdicke.
Parasternale lange Achse: Deutlich sichtbar die linksventrikuläre Hypertrophie mit erhöhter Septumdicke. Parasternale kurze Achse: Die Aufnahme zeigt ebenfalls eine linksventrikuläre Hypertrophie.
Parasternale kurze Achse: Die Aufnahme zeigt ebenfalls eine linksventrikuläre Hypertrophie. Gewebedoppler-Echokardiographie des Mitralanulus (Mitralring) mit nahezu normaler systolischer Funktion
Gewebedoppler-Echokardiographie des Mitralanulus (Mitralring) mit nahezu normaler systolischer Funktion



Bei vielen Morbus-Fabry-Patienten ist auch der rechte Ventrikel hypertroph (rechtsventrikuläre Hypertrophie, RVH). Die Herzkammer hat dabei eine normale Größe und die Systole ist ebenfalls normal; die diastolische Funktion ist massiv gemindert. Zwei Drittel der Patienten mit LVH zeigen auch das Symptom der RVH.[94][95] Die rechtsventrikuläre Hypertrophie ist eine wahrscheinliche Ursache dafür, dass Patienten mit einer guten linksventrikulären Herzfunktion eine geringe körperliche Ausdauer haben und unter Organomegalie (Organvergrößerung) sowie Lymphödemen leiden.[7][96]
Bedingt durch die Schädigung der Herzfunktion weisen die Elektrokardiogramme (EKG) von adulten Morbus-Fabry-Patienten mit der klassischen Ausprägung der Erkrankung charakteristische Veränderungen auf.[7]
- Elektrokardiogramme (EKG) von Patienten mit Morbus Fabry
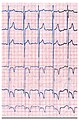 Das EKG eines Morbus-Fabry-Patienten zeigt eine linksventrikuläre Hypertrophie mit erhöhtem Sokolow-Lyon-Index, reduzierter ST-Strecke und negativen T-Wellen in den linken EKG-Ableitungen.
Das EKG eines Morbus-Fabry-Patienten zeigt eine linksventrikuläre Hypertrophie mit erhöhtem Sokolow-Lyon-Index, reduzierter ST-Strecke und negativen T-Wellen in den linken EKG-Ableitungen. Ein 24-Stunden-EKG wird häufig vor und nach einer Enzymersatztherapie durchgeführt, wenn der Patient über Herzrhythmusstörungen oder Herzklopfen berichtet.
Ein 24-Stunden-EKG wird häufig vor und nach einer Enzymersatztherapie durchgeführt, wenn der Patient über Herzrhythmusstörungen oder Herzklopfen berichtet.


Zerebrovaskuläre Schädigungen
Auf die frühen, meist in der Jugend auftretenden, Symptome der peripheren Neuropathien folgen im Erwachsenenalter häufig zerebrovaskuläre Erkrankungen und autonome Dysfunktionen (Erkrankungen beziehungsweise Funktionsstörungen des vegetativen Nervensystems). Einige der besonders ungünstigen neurologischen Wesensmerkmale von Morbus Fabry werden durch zerebrale, an mehreren Stellen auftretende (multifokale) Durchblutungsstörungen in den kleinen Blutgefäßen verursacht.[97][98] Die zerebrovaskulären Veränderungen können zu einer Vielfalt unterschiedlicher Anzeichen und Symptomen führen. Die Bandbreite geht dabei von Kopfschmerzen und Schwindelgefühlen, über transitorische ischämische Attacken und ischämische Schlaganfälle,[97][98][99] bis zu vaskulärer Demenz.[7][100][101]
Die Prävalenz für einen Hirninfarkt liegt bei männlichen Patienten bei etwa 6,9 % und bei weiblichen bei 4,3 %. Sie ist wesentlich höher als in der Gesamtbevölkerung. Das mittlere Alter beim ersten Hirninfarkt beträgt bei Männern mit Morbus Fabry etwa 39 und bei Frauen 46 Jahre. Nicht selten ist ein Hirninfarkt die erste Manifestation des Morbus Fabry.[98] In den meisten Fällen wird der Hirninfarkt durch kleine Blutgefäße ausgelöst. Daneben sind noch Dolichoektasien (syn. dilatative Arteriopathien, Arterienaufweitungen) der vertebrobasilären Zirkulation als Auslöser beschrieben.[7][99][102] Die Thrombenbildung wird möglicherweise durch eine gesteigerte Adhäsion von neutrophilen Granulozyten und Monozyten an den Endothelien[103] oder durch eine lokal erhöhte Durchblutung (Hyperperfusion) gefördert.[104][105][106] Der Serumspiegel des Enzyms Myeloperoxidase ist bei Männern mit Morbus Fabry ein Biomarker für das Risiko eines vaskulopathisch bedingten Vorfalls.[7][107]
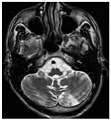 Die axiale MRT des Gehirns eines 27-jährigen Morbus-Fabry-Patienten mit ischämischem Schlaganfall zeigt den Schlaganfall in der linken zerebellären Hemisphäre. Der Patient wies sonst keine Symptome der Erkrankung auf.
Die axiale MRT des Gehirns eines 27-jährigen Morbus-Fabry-Patienten mit ischämischem Schlaganfall zeigt den Schlaganfall in der linken zerebellären Hemisphäre. Der Patient wies sonst keine Symptome der Erkrankung auf.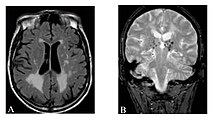 Hyperintensitäten der weißen Hirnsubstanz, lakunare Hirninfarkte und Mikroblutungen.
Hyperintensitäten der weißen Hirnsubstanz, lakunare Hirninfarkte und Mikroblutungen.
A) Die axiale MRT zeigt vielfältige Läsionen der weißen Hirnsubstanz in der zerebralen Hemisphäre eines 53-jährigen männlichen Patienten mit einem Fazekas-Score von 3.
B) Lakune und Mikroblutungen bei demselben Patienten. Die T1-gewichteten sagittalen (A) und axialen (B) MRTs zeigen ein symmetrisch hohes Signal im Thalamus (das sogenannte Pulvinar Sign) eines 66-jährigen männlichen Patienten. (C) und (D), ebenfalls T1-gewichtet, zeigen das Pulvinar Sign bei einem 42-jährigen Patienten.
Die T1-gewichteten sagittalen (A) und axialen (B) MRTs zeigen ein symmetrisch hohes Signal im Thalamus (das sogenannte Pulvinar Sign) eines 66-jährigen männlichen Patienten. (C) und (D), ebenfalls T1-gewichtet, zeigen das Pulvinar Sign bei einem 42-jährigen Patienten. Die Time-of-Flight-Magnetresonanzangiographien von vier Morbus-Fabry-Patienten zeigen erweiterte (ekstatische) Blutgefäße (Dolichoektasien der vertebrobasilären Zirkulation).
Die Time-of-Flight-Magnetresonanzangiographien von vier Morbus-Fabry-Patienten zeigen erweiterte (ekstatische) Blutgefäße (Dolichoektasien der vertebrobasilären Zirkulation).




Weitere Spätfolgen



Die an Nieren, Herz und Gehirn hervorgerufenen Schäden stellen den wesentlichen Anteil an der Mortalität des Morbus Fabry. Andere Spätfolgen sind klinisch relevant, leisten aber keinen oder nur einen geringen Beitrag zur Mortalität der Erkrankung. So sind beispielsweise Schäden am Gehör- und Gleichgewichtsorgan weit verbreitet. 80 % der männlichen und 77 % der weiblichen Patienten weisen einen progressiven Verlust des Gleichgewichtssinns auf.[108] Die Funktion des Gleichgewichtsorgans lässt sich mittels Kopfimpulstest überprüfen. Bei hemizygoten Patienten mit klassischem Verlauf der Erkrankung sind progressiver Hörverlust und plötzliche Taubheit ausgesprochen häufig.[109] Die Akkumulation von α-Galactosidase A kann darüber hinaus zu Tinnitus und Vertigo führen.[110][111]
Die Atemwege sind bei vielen Morbus-Fabry-Patienten ebenfalls von der Erkrankung betroffen. Atembeschwerden (Dyspnoe), chronischer Husten, sowie Fiepen und Giemen ist bei beiden Geschlechtern weit verbreitet.[112][113] Atemwegsobstruktionen weisen einer Studie zufolge 61 % der männlichen und 26 % der weiblichen Patienten auf.[7][114][115]
Veränderungen am Skelett, die im Wesentlichen die Knochendichte betreffen, sind auch ein häufiges Spätsymptom des Morbus Fabry. In einer Studie wurde bei 88 % der Patienten mit einem Durchschnittsalter von 31 Jahren und klassischem Verlauf der Erkrankung, mit Hilfe der Dual-Röntgen-Absorptiometrie (DXA) entweder eine Osteopenie oder das fortgeschrittene Stadium, eine Osteoporose, diagnostiziert.[116][117] In einer nachfolgenden größeren Studie wurde bei etwa 50 % der Morbus-Fabry-Patienten eine Osteopenie festgestellt.[118] Die reduzierte Knochendichte kann zu Spontanfrakturen führen.[119]
Diagnose
Bedeutung einer frühen Diagnose
Eine möglichst frühzeitige Diagnosestellung ist bei Morbus Fabry aus mehreren Gründen wichtig. Zum einen gibt es seit 2001 die Möglichkeit, die Krankheit kausal zu behandeln. Die Lebensqualität der Betroffenen kann dadurch erheblich verbessert werden und die Organschädigungen zumindest reduziert oder verzögert werden. Andererseits kann bei Familienangehörigen die genetische Veranlagung für die Krankheit noch vor dem Auftreten erster Symptome erkannt werden. In solchen Fällen ist eine Überwachung der Krankheitsentwicklung und frühzeitige Therapie möglich, bevor die Erkrankung symptomatisch wird.[46]
Fehldiagnosen
Aufgrund der Seltenheit der Erkrankung wird der Morbus Fabry von den meisten Kinderärzten und Internisten falsch diagnostiziert und entsprechend falsch behandelt. Eine Studie aus dem Jahr 2010 analysierte die Krankengeschichten von 45 Patienten mit Morbus Fabry. Die meisten Patienten klagten in ihrer Jugend über neuropathische Schmerzen als erstes Krankheitssymptom – es wurde in den meisten Fällen als „rheumatisches Fieber“ fehldiagnostiziert. Sieben Patienten wurden über Jahre mit Penicillin behandelt. Bei zehn Patienten mit Bauchschmerzen wurde eine Lebensmittelvergiftung oder „unspezifischer Schmerz“ diagnostiziert. Das Erstsymptom Anhidrose konnte keiner Ursache zugeordnet werden und Angiokeratome wurden als Petechien gedeutet. Im Durchschnitt dauerte es 19,7 Jahre, bis die korrekte Diagnose ‚Morbus Fabry‘ gestellt wurde.[120] In einer früheren Studie mit 366 Patienten betrug diese Zeitdifferenz bei Männern 13,7 und bei Frauen 16,3 Jahre.[121] Eine britische Studie aus dem Jahr 2001 ermittelte bei männlichen Patienten ein mittleres Alter von 22 Jahren für die Erstdiagnose, die mit einem durchschnittlichen Abstand von 8 Jahren nach den ersten Symptomen gestellt wurde.[122]
In dem langen Zeitraum zwischen ersten Symptomen und korrekter Diagnosestellung haben viele Patienten eine lange und frustrierende Odyssee von Arzt zu Arzt hinter sich.[9][123] In den meisten Fällen erfolgt die korrekte Diagnosestellung eher zufällig bei einem Augenarzt über die Cornea verticillata (Vortexkeratopathie) oder beim Hautarzt über die Angiokeratome.[122]
Korrekte Diagnose
Das klinische Bild kann bei der klassischen Form des Morbus Fabry wesentliche Beiträge zu einer frühen korrekten Diagnosestellung liefern; speziell die Angiokeratome und die Vortexkeratopathie. Diagnosesicherheit liefert bei männlichen Patienten die Bestimmung der Aktivität der α-Galactosidase A, aus dem Plasma oder aus Leukozyten, mit einem Enzymassay.[124] Die Bestimmung aus dem Plasma kann manchmal zu einer Fehldiagnose des Morbus Fabry führen, weshalb die Kontrolle des Ergebnisses mit der Aktivitätsbestimmung über die Leukozyten empfohlen wird.[125] Bei weiblichen Patienten ist diese Methode oft nicht ausreichend. Sie versagt bei über 30 % der Morbus-Fabry-Patientinnen, da sie eine für den Test zu hohe Restaktivität des Enzyms aufweisen.[126] Deshalb sollte bei allen Patientinnen, bei denen der Verdacht auf Morbus Fabry besteht, die Diagnose mittels Genotypisierung erfolgen.[20]
Das Gen wird sequenziert und mit bekannten GLA-Mutationen abgeglichen. Ergänzend ist die Bestimmung des Biomarkers Lyso-Gb3 sinnvoll. Bei unklarer Enzymaktivität kann bei Personen mit unspezifischen Morbus Fabry-Symptomen (LVH oder CKD u. a.) ein Lyso-Gb3-Wert von höher als 1,3 nmol/l auf Morbus Fabry hindeuten.[127] Sowohl für die Messung der Enzymaktivität und Lyso-Gb3 als auch für die genetische Analyse steht heute ein einfach in den Praxisalltag integrierbarer Trockenbluttest (Dried Blood Spot, DBS) zur Verfügung: Dafür werden einige Tropfen Blut auf eine Trockenblutkarte aufgetropft. Nachdem sie getrocknet sind, wird die Karte per Post an ein spezialisiertes Labor geschickt. Dort wird das Blut wieder aus der Filterkarte herausgelöst und für weitere Tests aufbereitet. Lyso-GL-3 ist ein guter Marker, um einen klassischen Morbus Fabry sicher auszuschließen. Bei Personen mit einer unsicheren GLA-Bedeutung, die eine unspezifische Morbus-Fabry-Symptomatik zeigen (LVH oder CKD u. a.) und keine charakteristischen, phänotypischen oder biochemischen Merkmale des klassischen Morbus Fabry aufweisen, sollte man bei erhöhten Lyso-GL-3-Werten > 1,3 nmol/l an Morbus Fabry denken.[127]
Die Plasma-[39] und Urinspiegel[128][129][130][131] von Gb3 können prinzipiell auch für die Diagnosestellung herangezogen werden. Die Urinwerte sind in ihrer Aussagekraft bei männlichen und weiblichen Patienten zwar zuverlässiger als die Plasmawerte; einige Patienten mit der Spätform des Morbus Fabry oder mit bestimmten Mutationsformen (beispielsweise p.Asn215Ser) weisen jedoch normale Gb3-Konzentrationen im Urin auf.[7][41][132]
Differentialdiagnostisch abzugrenzen ist das Cobb-Syndrom.
Pränatale Diagnose
Die Diagnose Morbus Fabry ist vorgeburtlich, das heißt pränatal, möglich. Dazu kann die biochemische oder die molekulare Pränataldiagnostik verwendet werden. Bei ersterer kann die Aktivität der α-Galactosidase A von Chorionzotten entweder direkt oder in einer Zellkultur gemessen werden. Die Entnahme per Chorionzottenbiopsie ist in der 10. Schwangerschaftswoche möglich. Die Diagnose von kultivierten amniotischen Zellen (Zellen im Fruchtwasser), die per Amniozentese aus dem Fruchtwasser entnommen werden, ist um die 14. Schwangerschaftswoche möglich. Aufwändiger ist die Bestimmung des Genotyps mittels DNA-Analyse (Gentest). Eine genetische Beratung wird üblicherweise vor der pränatalen Diagnostik durchgeführt. Aus ethischen Gründen wird die pränatale Diagnostik bei Morbus Fabry, speziell bei weiblichen Feten, sehr kontrovers diskutiert. Mit der Verfügbarkeit der Enzymersatztherapie hat sich diese Diskussion auf männliche Feten ausgeweitet. Einige Autoren empfehlen die pränatale Diagnostik grundsätzlich nur bei männlichen Feten. Die Geschlechtsbestimmung des Fetus ist in der 9. bis 11. Schwangerschaftswoche aus dem Blut der Mutter möglich.[7]
Die Präimplantationsdiagnostik ist prinzipiell möglich und wurde auch schon durchgeführt. Es gibt dazu bisher (Stand: September 2011) noch keine Veröffentlichungen.[133]
Neugeborenenscreening
Morbus Fabry ist derzeit kein Bestandteil der Neugeborenenscreenings in Deutschland und Österreich.[134] Es gilt die Prämisse, dass ein Screening nach einer bestimmten angeborenen Erkrankung nur dann sinnvoll ist, wenn es auch eine Behandlungsoption dafür gibt. Mit der Verfügbarkeit der Enzymersatztherapie ist diese Prämisse bei Morbus Fabry hinfällig.[135] Aus Trockenblutproben (dry blood spots, DBS) kann mittels HPLC-MS eine schnelle und relativ kostengünstige Analyse auf mehrere lysosomale Speicherkrankheiten erfolgen.[136] Mehrere groß angelegte Studien sind derzeit zur Überprüfung der Zuverlässigkeit des Verfahrens am Laufen.[137] Plasma-Lyso-GL-3 kann einfach im Trockenbluttest gemessen werden und bietet sich als effektiver und spezifischer Biomarker für das Screening von Neugeborenen mit Verdacht auf Morbus Fabry an, noch ehe erste Symptome sichtbar werden.[138]
Therapie

In Braun bei einem Morbus-Fabry-Patienten. Im Vergleich dazu in Grün ein gesunder Patient. In Gelb die grundsätzlichen Therapieansätze. In Rot die pathologischen Prozesse. (Grafik nach[32])
Die Entwicklung von wirksamen Behandlungsmethoden, insbesondere von Wirkstoffen, ist beim Morbus Fabry – wie bei allen lysosomalen Speicherkrankheiten – ausgesprochen schwierig. Einerseits gibt es wegen der niedrigen Inzidenz nur sehr wenige Patienten für die Durchführung von klinischen Studien und andererseits sind die Anforderungen bezüglich der Arzneimittelsicherheit bei der Einnahme über lange Zeiträume sehr hoch. Die Medikamente sind lebenslang und idealerweise vor dem Auftreten der ersten Symptome, also von weitgehend gesunden Patienten, einzunehmen.[139] Bedingt durch die Seltenheit der Erkrankung ist der Markt für ein entwickeltes Medikament ausgesprochen klein. Die in der Pharmaindustrie üblichen hohen Entwicklungskosten verteilen sich somit über eine kleine Anzahl von Patienten, was sehr hohe Behandlungskosten pro Patient zur Folge hat.
Bis zum Jahr 2001 konnten Morbus-Fabry-Patienten nur symptomatisch beziehungsweise palliativ behandelt werden. Bis zu diesem Zeitpunkt bestand die Behandlung im Wesentlichen in der Vermeidung von schmerzauslösender Reizen, wie beispielsweise Stress, körperliche Anstrengung, Hitze, Sonnenlicht und starken Temperaturänderungen. Gegen die Schmerzen gab es hochdosierte Analgetika. Mit einer vermehrten Flüssigkeitszufuhr bei heißem Wetter und der Vermeidung körperlicher Belastung wurde der Anhidrose entgegengewirkt. Eine fettarme Diät und Medikamente dienten der Linderung von Magen-Darm-Beschwerden. Nierenschonkost wurde bei leichter Proteinurie verordnet. Zur Prävention von Schlaganfällen wurden gerinnungshemmende Medikamente verschrieben.[140] Terminales Nierenversagen wurde – wie heute noch – per Nierenersatztherapie (Dialyse oder Nierentransplantation) behandelt.
Enzymersatztherapie
Die Enzymersatztherapie (ERT) ist derzeit (Stand Mai 2023) die einzige Möglichkeit zur ursächlichen Behandlung (kausale Therapie) des Morbus Fabry.[75] Für Morbus-Fabry-Patienten in der Europäischen Union gibt es zur Behandlung der Krankheit Agalsidase alfa und Agalsidase beta. Beides sind biotechnologisch hergestellte Varianten der α-Galactosidase A. Für US-amerikanische Patienten steht seit April 2003 Agalsidase beta zur Verfügung. Agalsidase alfa hat bis heute (Stand September 2011) in den Vereinigten Staaten keine Zulassung.[141] Außer in den 27 EU-Staaten ist es in insgesamt 45 Staaten, darunter Kanada, Japan, Brasilien und China zugelassen (Stand Mai 2011).[142] Zudem ist seit 2023 Pegunigalsidase alfa zugelassen.
Agalsidase alfa und beta
Beide Arzneistoffe werden gentechnisch hergestellt. Während bei Agalsidase alfa eine humane Fibroblastenzelllinie das Enzym produziert,[143] kommen bei Agalsidase beta Ovarzellen des Chinesischen Hamsters (CHO-Zellen) zum Einsatz. Agalsidase beta ist ein chimäres Protein. In ihrer Aminosäuresequenz sind die beiden Enzyme identisch, unterscheiden sich aber – bedingt durch die unterschiedliche Form der Proteinexpression bei der Produktion – geringfügig in der Art der Glykosylierung. Die Unterschiede bestehen im Wesentlichen im Anteil an Sialinsäuren und Mannose-6-phosphat. Agalsidase beta hat einen höheren Anteil an vollständig sialylierten Oligosacchariden und einen höheren Phosphorylierungsgrad. Bei in-vitro-Versuchen konnte bei Agalsidase beta eine erhöhte Bindung am Mannose-6-phosphat-Rezeptor und eine höhere Aufnahme in Fabry-Fibroblasten festgestellt werden. In vivo lässt sich dagegen kein funktioneller Unterschied zwischen beiden Enzymen ausmachen. Auch was den Grad der Antikörper-Kreuzreaktivität betrifft, sind sie nicht unterscheidbar.[144] Beide Enzyme müssen intravenös verabreicht werden. Enzyme, die systemisch wirken sollen, sind grundsätzlich oral nicht verfügbar, da sie im Darm weitgehend in ihre Bestandteile (Aminosäuren) zerlegt werden. Agalsidase alfa wird alle zwei Wochen infundiert. Agalsidase beta wird im gleichen Rhythmus, aber anderer Dosierung, verabreicht.[75][145]
Wirksamkeit

Aufgrund der langsamen und über Jahrzehnte gehenden Progression des Morbus Fabry, sowie seiner Seltenheit, liegen bisher nur wenige gesicherte Daten über die langfristigen Behandlungserfolge vor. Bei den wesentlichen messbaren Krankheitsparametern konnte in einer vergleichenden Studie über einen Zeitraum von 24 Monaten kein Unterschied zwischen Agalsidase alfa und Agalsidase beta festgestellt werden.[34] Vor allem in den Endothelzellen kann die Enzymersatztherapie die Akkumulation von Gb3 im Lysosom deutlich reduzieren. Dies ist für die Behandlung der primären Morbiditätsfaktoren (Nierenversagen, kardiale und zerebrovaskuläre Erkrankungen) von großer Wichtigkeit. Allerdings ist die Wirksamkeit der Therapie bei Patienten mit fortgeschrittenen Symptomen eher bescheiden.[34][146] Eine möglichst frühzeitige Behandlung ist daher für den Therapieerfolg besonders wichtig. Der Abbau von Gb3 ist bei der Enzymersatztherapie abhängig vom Zelltyp. In den Nieren wird Gb3 – außer in den vaskulären Endothelien – auch in den Mesangialzellen des Glomerulums und Zellen im Interstitium des Nierenkortex abgebaut. Deutlich schlechter wird das Gb3 in der glatten Muskulatur der Arteriolen und kleinen Arterien, den Podozyten und im Epithel des distalen Tubulus reduziert.[147][148]
In klinischen Studien konnte die Verabreichung von Agalsidase alfa die Schmerzen mindern und die linksventrikuläre Hypertrophie reduzieren. Die Nierenfunktion wurde stabilisiert, das Gehör und die Fähigkeit zur Schweißabsonderung wurden verbessert. Insgesamt wurde die Lebensqualität deutlich gesteigert. Bei Patienten mit chronischem Nierenversagen konnte die Progression zum terminalen Nierenversagen verzögert werden.[149][150] Bei der Behandlung mit Agalsidase beta wurde eine Abreicherung von Gb3 in verschiedenen Zellen nachgewiesen. Auch die Re-Akkumulation von Gb3 bleibt aus. Die Verabreichung von Agalsidase beta reduziert das Risiko eines schwerwiegenden klinischen Ereignisses (beispielsweise Myokardinfarkt, terminales Nierenversagen oder Tod) signifikant.[151]
Nebenwirkungen
Etwa die Hälfte aller Patienten zeigt leichte bis mittelschwere infusionsbedingte Reaktionen, die ihren Höhepunkt zwischen der fünften und achten Infusion haben. Daneben zeigen einige Patienten Fieber und Schüttelfrost. Diese Nebenwirkungen sind von kurzer Dauer, nicht schwerwiegend und können konservativ behandelt werden.[150] Nach drei bis fünf Jahren Behandlungsdauer zeigen nur noch 10 bis 20 % der Patienten infusionsbedingte Reaktionen, wobei sich offensichtlich eine Infusionstoleranz entwickelt.[146][152] Die genaue Ursache der infusionsbedingten Reaktionen ist noch unbekannt. Man vermutet eine spezifische IgG-Antikörper-Bildung auf das infundierte Enzym. Bei 24 % der Patienten, die Agalsidase alfa erhalten, und bei 88 % der Patienten mit Agalsidase beta wurde eine IgG-Serokonversion festgestellt.[7] Die Bildung von Antikörpern kann bei Patienten auftreten, die keinerlei Restaktivität an α-Galactosidase aufweisen. Für ihr Immunsystem ist α-Galactosidase „neu“ und „fremdartig“.[153] Die Bildung von Antikörpern gegen die beiden Agalsidase-Präparate reduziert unmittelbar deren Wirksamkeit.[154][155][156] Eine multizentrische Untersuchung zeigte jedoch, dass ein „Überspritzen“ von Antikörpern durch entsprechende Enzymmengen in weiterhin niedrigen Spiegeln des Krankheitsmarkers Lyso-Gb3 resultiert.[157]
Wettbewerbssituation und Produktionsprobleme
Shire Pharmaceuticals erzielte 2015 mit Replagal einen Umsatz von 326 Millionen[158] und Genzyme mit Fabrazyme 188 Millionen US-Dollar. Zwischen beiden Unternehmen herrscht auf dem europäischen Markt ein harter Wettbewerb, während in den Vereinigten Staaten nur Fabrazyme zugelassen ist. Genzyme verlor in Europa Marktanteile, weil im Rahmen eines Lieferengpasses Patienten von der vollen Dosis Fabrazyme auf niedrigere Dosierungen oder Replagal umgestellt wurden. Als es zu diesem Lieferengpass mit Fabrazyme kam, wurde ein Großteil der amerikanischen Ware für den Export nach Europa allokiert. Dies führte dazu, dass US-Patienten entweder eine niedrigere Dosierung oder überhaupt kein Arzneimittel zur Behandlung zur Verfügung stand, während in Europa etwa 400 Patienten mit der vollen Dosis ausreichend versorgt wurden.[159][160]
Zu den Lieferschwierigkeiten kam es erstmals 2009 durch eine Kontamination mit eingeschleppten Caliciviren (Typ: Vesivirus 2117) in den weltweit einzigen Bioreaktoren zur Produktion von Fabrazyme in Allston (Massachusetts).[161] Das Virus ist zwar für Menschen ungefährlich, hatte aber erheblichen Einfluss auf die Produktionsausbeute. Die Qualität des Produktes war dagegen nicht betroffen. Die Produktionsanlage stand zudem längere Zeit wegen Desinfektionsmaßnahmen still.[162] Genzyme wurde mit einem Bußgeld in Höhe von 175 Millionen US-Dollar belegt.[159]
Im August 2010 verlangten drei US-amerikanische Morbus-Fabry-Patienten in einer Petition von den National Institutes of Health (NIH), dass Genzyme anderen Unternehmen die Enzymersatztherapie auf der Basis der March-in Rights ermöglicht. Die March-in Rights sind besondere Eingriffsrechte des Staates, die dann bestehen, wenn Forschung und Entwicklung staatlich finanziert wurden. Der Bayh–Dole Act von 1980 erlaubt unter anderem Universitäten, dass sie die Ergebnisse staatlich finanzierter Forschung selbst vermarkten können.[163] Im Gegenzug ermöglicht der Bayh-Dole Act den finanzierenden staatlichen Stellen, dass sie die Exklusivrechte am geistigen Eigentum (in diesem Fall Patente) außer Kraft setzen können, beispielsweise wenn Menschen in Lebensgefahr sind.[142][164] Agalsidase beta wurde an der Mount Sinai School of Medicine mit NIH-Geldern entwickelt und mit zwei Patenten (US 5.356.804 und US 5.580.757) geschützt. Genzyme ist exklusiver Lizenznehmer beider Patente. Die Petition der Patienten wurde von den NIH im Dezember 2010 abgelehnt.[165] Als Hauptgrund führt die Behörde an, dass eine weitere Lizenzierung an Dritte die von den Antragstellern angeführten Probleme – im Wesentlichen der akute Versorgungsengpass – nicht lösen würde. Es würden von jeder Lizenzvergabe bis zur Verfügbarkeit des Arzneimittels von einem weiteren Hersteller Zeiträume von mehreren Jahren vergehen, da aufwändige klinische Studien und Zulassungsprozesse notwendig seien. Zudem verspreche Genzyme, noch im ersten Halbjahr des Jahres 2010 wieder die volle Produktionskapazität zu erreichen.[164][166]
Die Produktionsprobleme von Fabrazyme bestanden noch im September 2011.[167] Im ersten Quartal 2012 wurde die Produktion an einer neuen Betriebsstätte in Framingham (Massachusetts) aufgenommen, die Anlage wurde von der FDA genehmigt.[168] 100 US-amerikanische Patienten erhalten, im Rahmen einer von der FDA genehmigten klinischen Studie, derzeit (Stand: September 2011) das Konkurrenzprodukt Replagal.[159]
Nachdem die US-Aufsichtsbehörden grünes Licht für die Herstellung von Fabrazyme am Standort Framingham (Massachusetts) gegeben hatten, begann Sanofi Genzyme Corp. mit der Auslieferung.[169] Zuvor hatten die FDA und die EMA im Januar 2012 die Anlage in Framingham für die Produktion von Fabrazyme genehmigt.[169]
Im März 2012 wurde bekannt, dass Shire Pharmaceuticals den Zulassungsantrag für Replagal in den USA zurückgezogen hat.[170] Trotz einer von der US Behörde FDA ausgesprochenen Empfehlung und trotz eines von der FDA vergebenen Fast Track Statuses kam es zu keiner Zulassung auf dem US-Markt.[170][171]
Die Patente der Mount Sinai School of Medicine 5.356.804 liefen am 27. September 2015 für den US-Markt sowie im August 2016 für den europäischen Markt ab.[172]
Therapiekosten
Die Behandlungskosten liegen in Deutschland pro Patient und Jahr bei etwa 250.000 €, unabhängig davon ob Agalsidase alpha oder beta verwendet wird.[173] Eine Ampulle mit 3,5 mg Agalsidase alfa, für die Dosierung 0,2 mg pro kg Körpergewicht, kostet in Frankreich 1685 €. Für eine Ampulle mit 35 mg Agalsidase beta, mit der Dosierung 1 mg pro kg Körpergewicht, fallen 3370 € an. Pro Patient ergeben sich dadurch identische jährliche Behandlungskosten, die in Frankreich bei 161.781 € für einen 70 kg schweren Patienten liegen (Stand: 2009).[7] Da die Enzymersatztherapie die derzeit einzige kausale therapeutische Option beim Morbus Fabry ist, werden die Kosten dafür in Deutschland von den gesetzlichen Krankenkassen erstattet und nicht auf das Verschreibungsbudget der Praxis des verschreibenden Arztes angerechnet.[174]
Pegunigalsidase alfa
Pegunigalsidase alfa (prh-alpha-Gal-A) ist eine pegylierte rekombinante Form der menschlichen α-Galaktosidase A, die sowohl in den USA als auch in der EU im Mai 2023 für die langfristige Enzymersatztherapie bei erwachsenen Patienten (Elfabrio) zugelassen wurde.[175][176] Pegunigalsidase alfa wird in Zellen der Tabakpflanze (Nicotiana tabacum-BY-2-Zellen) mittels rekombinanter DNA-Technologie hergestellt. In Studien wurde bei manchen Patienten eine geringere Affinität von Anti-Drug-Antikörpern gegenüber Pegunigalsidase alfa im Vergleich zu Agalsidase alfa festgestellt.[177] Pegunigalsidase alfa soll sich günstiger bezüglich der Verschlechterung der Nierenfunktion auswirken.[178] Die Daten aus den Zulassungsstudien ließen keine Aussage über einen möglichen Vorteil von Pegunigalsidase alfa in Bezug auf das Immunogenitäts- und Nebenwirkungsprofil im Vergleich zu Agalsidase beta zu. Trotz der deutlich längeren Plasmahalbwertszeit entspricht das Dosierungsintervall (1× alle zwei Wochen) demjenigen von Agalsidase alfa und beta.[179]
Chaperon-Therapie

Ein Großteil der Mutationen auf dem GLA-Gen ist vom Typ missense. Unter den Genprodukten befinden sich sehr viele Enzymvarianten, die in aufgereinigter Form in ihrer Aktivität vergleichbar mit dem Wildtyp der α-Galactosidase A sind, aber eine geringere thermische und pH-Stabilität aufweisen. Diese mutierten Varianten der α-Galactosidase A werden – wie alle neu synthetisierten Proteine – im endoplasmatischen Retikulum (ER) einer „Qualitätskontrolle“ unterzogen (Proteinqualitätskontrolle, protein quality control machinery). Sie sorgt dafür, dass nur richtig gefaltete und modifizierte Proteine an ihren Bestimmungsort gelangen. Proteine, die diese Qualitätskontrolle nicht bestehen, werden aus dem endoplasmatischen Retikulum in das Zytosol transportiert und im Proteasom zerlegt. Dieser Vorgang wird ER-assoziierte Proteindegradation (ERAD, Endoplasmic Reticulum Associated Protein Degradation) genannt. Die durch die Mutation falsch gefalteten Varianten der α-Galactosidase A werden im Rahmen der Qualitätskontrolle ausgesondert und zerlegt.[181]
Dieser Prozess bietet jedoch eine Möglichkeit zur therapeutischen Intervention. Mit der Hilfe von pharmakologischen Chaperonen oder chemischen Chaperonen kann die Fehlfaltung der mutierten Enzyme korrigiert werden. Bei den pharmakologischen Chaperonen handelt es sich um kleine Moleküle (small molecules) die als Faltungs-Templat des Enzyms dienen. Dadurch wird die Faltungsdynamik des Proteins in Richtung der richtigen Konformation verschoben und stabilisiert. Mit der korrekten Tertiärstruktur wird die Qualitätskontrolle im ER „bestanden“.[182][183] Der stabile Chaperon-Protein-Komplex wird durch Vesikel des endoplasmatischen Retikulums in den Golgi-Apparat und dann in das Lysosom transferiert. Dort wird das pharmakologische Chaperon durch das natürliche Substrat (Gb3) ersetzt.[184] Die Dissoziation des Chaperon-Protein-Komplexes wird dabei durch die hohe Konzentration an Gb3 und den niedrigen pH-Wert im Lysosom begünstigt.[185]
Der Iminozucker 1-Deoxygalactonojirimycin (DGJ), internationaler Freiname Migalastat, ist ein Beispiel für ein pharmakologisches Chaperon. Es ist ein Analogon der terminalen Galactose von Gb3 und ein reversibler Inhibitor der α-Galactosidase A.[186] In einer Vielzahl von präklinischen Versuchen konnte gezeigt werden, dass Migalastat in der Lage ist, die Aktivität mutierter Varianten der α-Galactosidase A zu erhöhen.[184] Dadurch konnte beispielsweise in Fabry-Mäusen die Akkumulation von Gb3 signifikant reduziert werden. Als kleines Molekül hat Migalastat eine sehr breite Bioverteilung im Organismus und kann beispielsweise das Zentralnervensystem erreichen und die Blut-Hirn-Schranke überwinden.[187] Darüber hinaus ist es oral verfügbar.[188]
Migalastat ist seit 2016 in der Europäischen Union zugelassen.[189][190] Bisher ist noch unklar welcher Anteil an Morbus-Fabry-Patienten mit dieser Therapieform zukünftig behandelt werden könnten, da sich nicht alle der bisher bekannten über 500 Mutationsformen der α-Galactosidase A mit pharmakologischen Chaperonen aktivieren beziehungsweise korrekt entfalten lassen. In einer Untersuchung von 299 Mutationsformen werden 40 davon als potenziell mit pharmakologischen Chaperonen therapierbar eingestuft. Speziell bei den Mutationen die einen nicht-klassischen Krankheitsverlauf bewirken, bestehen gute Erfolgsaussichten.[187] Die Analyse des Genotyps wird vor einem möglichen Therapiebeginn obligat sein.[191]
Begleitende Therapien
Speziell bei jungen Patienten reduziert die Enzymersatztherapie die neuropathischen Schmerzen recht erfolgreich.[192] Eine begleitende Schmerztherapie ist aber in vielen Fällen dennoch angezeigt. Zur Behandlung der neuropathischen Schmerzen bei Morbus Fabry wird Carbamazepin,[193] gegebenenfalls in Kombination mit Pregabalin[194] als Mittel der ersten Wahl empfohlen. Bei unerträglichen Schmerzkrisen können auch Opioide zur Anwendung kommen.[13][195][196] Gegen die gastrointestinalen Beschwerden werden Medikamente wie Metoclopramid empfohlen, die den Bewegungsstörungen im oberen Magen-Darm-Trakt (Motilitätsstörungen) entgegenwirken.[197]
Liegt eine erhöhte Eiweißausscheidung im Urin als Hinweis auf eine Nierenschädigung vor, kann durch zusätzliche Behandlung mit ACE-Hemmern oder AT1-Antagonisten, zwei verwandten Klassen blutdrucksenkender Medikamente, das Fortschreiten der Nierenschädigung verzögert werden.[198] Ist das Herz bereits geschädigt, so kann durch ACE-Hemmer der arterielle Gefäßwiderstand und damit der arterielle Blutdruck gesenkt werden. Dadurch wird auch die Vor- und Nachlast des Herzens bei einer Myokardinsuffizienz vermindert und das Herzminutenvolumen erhöht. Die Herzfrequenz und der Sauerstoffbedarf des Myokards kann durch Betablocker reduziert werden. Herzrhythmusstörungen können beispielsweise mit Amiodaron korrigiert werden. Daneben bleiben noch chirurgische Maßnahmen, wie beispielsweise die Implantation eines Herzschrittmachers, eines Koronarstents, einer künstlichen Herzklappe oder eines Koronararterien-Bypasses.[199]
Mögliche zukünftige Therapieoptionen
Die Enzymersatztherapie kann in vielen Fällen die Progression des Morbus Fabry stoppen oder zumindest vermindern. Die Behandlung ist rein palliativ, das bedeutet, dass eine vollständige Heilung damit nicht möglich ist. Darüber hinaus sprechen nicht alle Patienten optimal auf diese Form der Behandlung an. Die Darreichungsform ist auf die intravenöse Applikation festgelegt. Die generell von Patienten bevorzugte orale Einnahme ist nicht möglich. Bei der Resorption im Magen-Darm-Trakt wird α-Galactosidase A in unwirksame Fragmente zerlegt. Ein Entwicklungsschwerpunkt sind daher oral verfügbare Arzneimittel, die in ihrer Wirksamkeit der Enzymersatztherapie mindestens gleichwertig sind oder aber diese therapeutisch zweckmäßig ergänzen können.
Auch die Enzymersatztherapie selbst wird weiterentwickelt. Ein möglicher künftiger Arzneistoff ist dabei eine modifizierte Form des Enzyms α-N-Acetylgalactosaminidase (NAGA). α-N-Acetylgalactosaminidase hat eine der α-Galactosidase A sehr ähnliche Struktur. In der aktiven Tasche unterscheiden sie sich nur in zwei Aminosäurenpositionen.[200] Durch gezielte Veränderungen (Protein-Engineering) an der aktiven Tasche dieses im Menschen vorkommenden Enzyms, ist es in der Lage Gb3 als Substrat aufzunehmen und wie α-Galactosidase A zu zerlegen. Der große Vorteil der modifizierten α-N-Acetylgalactosaminidase ist dabei, dass sie in Morbus-Fabry-Patienten nicht immunogen ist. Ihr Immunsystem „kennt“ α-N-Acetylgalactosaminidase.[7][153]
Substratreduktionstherapie

Während bei der Enzymersatztherapie des Morbus Fabry versucht wird die fehlende beziehungsweise defekte α-Galactosidase durch die Gabe von wirksamem künstlich hergestelltem Enzym zu ersetzen, um in den Zellen das Substrat Gb3 zerlegen zu können, geht man bei der Substratreduktionstherapie (SRT) einen anderen Weg. Hier versucht man das Substrat zu reduzieren und dadurch die Akkumulation von Gb3 in den Zellen zu unterbinden. Dies ist beispielsweise durch die Inhibierung des Enzyms Glucosylceramidsynthase (GCS, auch Ceramidglucosyltransferase genannt) möglich. GCS katalysiert den ersten Schritt der Synthese der Glycosphingolipide und dadurch auch die Synthese der nachfolgenden Moleküle, einschließlich Gb3. Im Modellorganismus Maus konnte die prinzipielle Wirksamkeit dieses therapeutischen Ansatzes gezeigt werden.[201][202] Die SRT könnte eine künftige ergänzende Behandlungsoption zur Enzymersatztherapie bei Morbus Fabry werden. Ein Beispiel eines Glycosyltransferase-Inhibitors ist Miglustat.[203] Dieser Arzneistoff ist zur Behandlung von zwei lysosomalen Speicherkrankheiten zugelassen: für die Niemann-Pick-Krankheit Typ C und Morbus Gaucher Typ 1. Bei letzterem allerdings nur bei Patienten, für die die ERT keine Behandlungsoption ist. Für die Therapie des Morbus Fabry ist Miglustat nicht zugelassen.[58] Das Nebenwirkungsprofil von Miglustat ist mit Durchfall und peripherer Neuropathie[204][205] für Morbus Fabry ungünstig und könnte die Symptome der Erkrankung weiter verstärken.[148] Die SRT ist im Fall des Morbus Fabry, bei dem ein Großteil der Patienten keinerlei Restaktivität des mutierten Enzyms aufweist, als Monotherapie eher unwahrscheinlich. Zukünftige SRT-Ansätze zielen daher eher auf eine Unterstützung der Enzymersatztherapie. Ein potenzieller Arzneistoff ist dabei Eliglustat.[148]
Knochenmarktransplantation
Im Modellorganismus Maus mit abgeschaltetem gal-Gen konnte nachgewiesen werden, dass mit einer Stammzelltransplantation aus dem Knochenmark von Mäusen des Wildtyps die Aktivität an α-Glaktosidase A ansteigt.[206][207] Dies kann mit einer reduzierten Konditionierung erzielt werden, bei der eine etwa 30%ige Genkorrektur offensichtlich ausreicht.[208] Der Eingriff und die Konditionierung sind mit einem wesentlich höheren Risiko als die Enzymersatztherapie behaftet. Es liegen noch keine Ergebnisse und Erfahrungen dieser Therapieform bei Morbus Fabry vor.
Gentherapie
Die Gentherapie ist eine vielversprechende zukünftige Behandlungsmöglichkeit, bei der die Hoffnung besteht, dass durch eine Einmalbehandlung eine Heilung erzielt werden kann. Dabei würde DNA beziehungsweise RNA mit dem genetischen Code für das GLA-Gen in die Körperzellen eingefügt werden.[209] Der therapeutische Ansatz wäre in der Kausalkette mutiertes Gen → defektes Enzym → mangelnde Funktion → Ablagerung von Gb3 → Morbus Fabry eine Stufe früher, als bei der Enzymersatztherapie. In präklinischen Versuchen an Fabry-Mäusen konnten mit verschiedenen Vektoren, vor allem viralen Vektoren, vielversprechende Ergebnisse erhalten werden.[210][211] Es ist bisher noch nicht abzusehen ob und wann diese Behandlungsmethode für Morbus-Fabry-Patienten zur Verfügung stehen wird.[140] Allgemein wurden die hohen Erwartungen in die Gentherapie, die vor allem um die Jahrtausendwende bestanden, in den letzten Jahren deutlich zurückgenommen. Bei einigen Varianten des Verfahrens, so bei der Verwendung von Retroviren für die Transduktion, besteht die Gefahr der Induktion von Krebs, durch die Inaktivierung von Tumorsuppressorgenen. Eine erfolgreiche somatische Gentherapie könnte einem behandelten Morbus-Fabry-Patienten zwar eine vollständige Heilung bringen, allerdings wäre in seinen Keimzellen weiterhin das defekte GLA-Gen. Seine Kinder hätten deshalb nach wie vor eine etwa 50%ige Wahrscheinlichkeit die Krankheit zu bekommen. Keimbahntherapien am Menschen, die eine Weitergabe des defekten Gens an die Nachkommen verhindern würden, sind in Deutschland und den meisten anderen Staaten verboten.[212]
Tiermodelle
Für die Entwicklung neuer Therapiemöglichkeiten des Morbus Fabry und zur Erforschung der Krankheit werden gentechnisch veränderte Organismen verwendet.[202] Es wurde bisher kein Großtier-Modell für den Morbus Fabry entdeckt, weshalb als Modellorganismus im Wesentlichen die a-gal-A-knockout-Maus (a-gal-A−/0-Maus oder ‚Fabry-Maus‘) verwendet wird. Bei dieser Knockout-Maus wird durch Gen-Knockout das gal-Gen abgeschaltet. Dies hat aber, im Vergleich zum Gendefekt beim Menschen, kaum Auswirkungen auf den Phänotyp. Die Mäuse sind selbst in einem Alter von 80 Wochen klinisch unauffällig, haben normale Blut- und Urinwerte und zeigen die gleiche Lebenserwartung wie der Wildtyp. Histopathologisch lässt sich die Akkumulation von Gb3 in der Leber und den Nieren nachweisen, die bei älteren Mäusen vom Genotyp a-Gal A−/0 morphologische Veränderungen im Gewebe bewirkt, aber zu keinen Läsionen führt.[206] Die Verfügbarkeit der Fabry-Maus hat einen wesentlichen Anteil an der Entwicklung der Enzymersatztherapie für Morbus Fabry, insbesondere in der präklinischen Phase.[202][213] Auch in präklinischen Studien zur Gen-, Chaperon- und Substratreduktionstherapie[148] werden a-Gal-A−/0-Mäuse verwendet.[214]
Prognose
Mit steigendem Alter nehmen die durch die Akkumulation von Gb3 ausgelösten Schäden an lebenswichtigen Organen immer weiter zu, bis diese Organe vollständig ihre Funktion verlieren. Terminale Niereninsuffizienz und lebensbedrohliche kardiovaskuläre oder zerebrovaskuläre Komplikationen begrenzen die Lebenserwartung von nicht-therapierten Patienten auf durchschnittlich etwa 50 und bei Patientinnen auf etwa 70 Jahre. Im Vergleich zur Gesamtbevölkerung entspricht dies einer Reduktion um 20 beziehungsweise 15 Jahre.[78][215]
In einer Studie waren die Haupttodesursachen Nierenversagen, zerebrovaskuläre Krankheiten (beispielsweise Hirnblutung oder Hirninfarkt) und Herzerkrankungen. Der Anteil an Todesfällen durch Nierenversagen nahm mit der Einführung der Dialyse und Transplantation ab. Die primäre Todesursache im Studienzeitraum von 2001 bis 2007 waren Herzerkrankungen; bei Männern zu 34 und bei Frauen zu 57 %. Die Ursache für die Verschiebung der Todesursachen wird in der verbesserten klinischen Versorgung der Patienten mit terminaler Niereninsuffizienz gesehen.[216]
Darüber, ob und gegebenenfalls in welchem Ausmaß die Enzymersatztherapie die Lebenserwartung der Patienten erhöhen kann, liegen noch keine statistisch signifikanten Daten vor.[7]
![Die Überlebensrate nach Kaplan-Meier für männliche Morbus-Fabry-Patienten im Vergleich zur männlichen Normalbevölkerung. Die mittlere Lebenserwartung liegt bei 50 Jahren, was einer Reduktion um etwa 20 Jahre entspricht.[122] Die Daten sind aus dem Jahr 2001, als noch keine Enzymersatztherapie verfügbar war.](https://upload.wikimedia.org/wikipedia/commons/thumb/3/37/Morbus_Fabry_survival_male.svg/214px-Morbus_Fabry_survival_male.svg.png) Die Überlebensrate nach Kaplan-Meier für männliche Morbus-Fabry-Patienten im Vergleich zur männlichen Normalbevölkerung. Die mittlere Lebenserwartung liegt bei 50 Jahren, was einer Reduktion um etwa 20 Jahre entspricht.[122] Die Daten sind aus dem Jahr 2001, als noch keine Enzymersatztherapie verfügbar war.
Die Überlebensrate nach Kaplan-Meier für männliche Morbus-Fabry-Patienten im Vergleich zur männlichen Normalbevölkerung. Die mittlere Lebenserwartung liegt bei 50 Jahren, was einer Reduktion um etwa 20 Jahre entspricht.[122] Die Daten sind aus dem Jahr 2001, als noch keine Enzymersatztherapie verfügbar war.![Die entsprechende Überlebensrate für weibliche Patienten. Die mittlere Lebenserwartung liegt bei 70 Jahren, was einer Reduktion um etwa 15 Jahre entspricht.[215] Auch diese Daten sind aus dem Jahr 2001.](https://upload.wikimedia.org/wikipedia/commons/thumb/8/8e/Morbus_Fabry_survival_female.svg/215px-Morbus_Fabry_survival_female.svg.png) Die entsprechende Überlebensrate für weibliche Patienten. Die mittlere Lebenserwartung liegt bei 70 Jahren, was einer Reduktion um etwa 15 Jahre entspricht.[215] Auch diese Daten sind aus dem Jahr 2001.
Die entsprechende Überlebensrate für weibliche Patienten. Die mittlere Lebenserwartung liegt bei 70 Jahren, was einer Reduktion um etwa 15 Jahre entspricht.[215] Auch diese Daten sind aus dem Jahr 2001.
![Die Überlebensrate nach Kaplan-Meier für männliche Morbus-Fabry-Patienten im Vergleich zur männlichen Normalbevölkerung. Die mittlere Lebenserwartung liegt bei 50 Jahren, was einer Reduktion um etwa 20 Jahre entspricht.[122] Die Daten sind aus dem Jahr 2001, als noch keine Enzymersatztherapie verfügbar war.](https://www.bahnsport-info.de/bahnsport-info-wikipedia/eisspeedway/?rdp_we_resource=https%3A%2F%2Fde.wikipedia.org%2Fwiki%2FDatei%3AMorbus_Fabry_survival_male.svg)
![Die entsprechende Überlebensrate für weibliche Patienten. Die mittlere Lebenserwartung liegt bei 70 Jahren, was einer Reduktion um etwa 15 Jahre entspricht.[215] Auch diese Daten sind aus dem Jahr 2001.](https://www.bahnsport-info.de/bahnsport-info-wikipedia/eisspeedway/?rdp_we_resource=https%3A%2F%2Fde.wikipedia.org%2Fwiki%2FDatei%3AMorbus_Fabry_survival_female.svg)
Medizingeschichte
Der Morbus Fabry wurde als eigenständiges Syndrom erst relativ spät entdeckt. Die ersten Veröffentlichungen über die Krankheit stammen von Johannes Fabry und William Anderson (beide im Jahr 1898). Die allgemeine mittlere Lebenserwartung lag 1820 bei etwa 30 Jahren und um 1900 bei ungefähr 40 Jahren. Im Vergleich dazu liegt heute die mittlere Lebenserwartung von männlichen Patienten mit Morbus Fabry bei etwa 45 bis 50 Jahren. Die Erkrankung ist somit nicht nur ausgesprochen selten, sondern war auch bei der allgemein kurzen Lebenserwartung klinisch unauffällig. Dermatologen konnten zum Ende des 18. Jahrhunderts Hautkrankheiten lediglich beschreiben. Die Bedeutung von Begleitsymptomen und die Verflechtung dieser untereinander war weitgehend unbekannt.
Die Phase der empirischen Dermatologie endete erst in den 1970er Jahren mit dem Aufkommen von mikrobiologischen und immunologischen Verfahren.[217]
Johannes Fabry und sein Patient

Der deutsche Dermatologe Johannes Fabry arbeitete zu dieser Zeit am Städtischen Krankenhaus Dortmund. Er veröffentlichte im Dezember 1898 im Archiv für Dermatologie und Syphilis den Artikel Ein Beitrag zur Kenntniss der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae).[218]
Fabry nannte in seinem Artikel die durch die Angiokeratome verursachten Hautläsionen Purpura papulosa haemorrhagica Hebrae. Diese Bezeichnung wählte Fabry,
„… weil wir vor Allem vermeiden müssen, zu der grossen und vielumstrittenen Lichengruppe noch ein neues Krankheitsbild hinzuzufügen; dann aber vor Allem, weil die Hebra’sche Bezeichnung in dem Worte ‚Purpura‘ zugleich das Genus trifft und das Epitheton die klinische Abart und Eigenthümlichkeit der Knötchenform und des papulösen Exanthems.“
Fabry war sich der Erstbeschreibung aber bewusst. Er schrieb: „Doch zuvor der genaue Krankenbericht des interessanten und, wie ich glaube, in der Literatur ohne ein Analogon dastehenden Falles.“[218]

Nachfolgend beschrieb er die Krankengeschichte von Emil Honke, einem 13-jährigen Jungen aus Langendreer. Dessen Eltern bemerkten bei ihm im Alter von neun Jahren kleine Knötchen in der Kniekehlengegend. Der Ausschlag breitete sich im Laufe der Jahre an der Rückseite der Oberschenkel bis zum Stamm hin aus. Mit zwölf traten die Knötchen dann auch an der linken Kniekehle auf. In den ersten Jahren blieben weitere Symptome aus, doch dann wurde Emil immer schwächlicher und appetitloser. Das Hautleiden selbst führte bei ihm zu keinen Beschwerden – kein Jucken, kein Stechen, kein Schmerz und kein Unbehagen. Die beiden Eltern – Anfang 40 – beschrieb Fabry als gesund. Der Großvater väterlicherseits verstarb im Alter von 49 Jahren an einem Nierenversagen.[217] Den Befund erstellte Fabry am 15. April 1897. Er behielt Emil mehrere Tage auf der Hautstation des städtischen Krankenhauses Dortmund und entnahm Gewebeproben der Haut. Fabry verschrieb ihm Eisentropfen und sah von einer örtlichen Behandlung ab. Die Knötchen schilderte Fabry als dunkelblau bis ins Schwarze gehend. Das Tastgefühl beim Streichen über die Haut der Brust beschrieb Fabry als „das typische Gefühl des Reibeisens“, ähnlich wie beispielsweise beim Morbus Darier.
Vor seiner Veröffentlichung präsentierte Fabry 1897 den bei Emil Honke erhaltenen histopathologischen Befund auf der Versammlung der Ärzte des Regierungsbezirks Arnsberg und in der dermatologischen Sektion der Braunschweiger Naturforscherversammlung.[219]

Nach 17 Jahren, am 11. Februar 1915, kam Emil Honke wegen seines Leidens wieder zu Fabry. Dieser hatte den Fall inzwischen völlig aus den Augen verloren. Honke hatte inzwischen als Anstreicher gearbeitet und dann drei Jahre als Bergmann.
„Wir hatten nun diesen Fall ganz aus dem Auge verloren und waren natürlich sehr überrascht, als [der] Patient sich in diesem Jahre erneut zur Untersuchung vorstellte. Aus der Anamnese ist hervorzuheben, daß das Hautleiden ihn nicht gehindert hat, seinem Beruf nachzugehen.“
Fabry veröffentlichte 1916 in einem Artikel über Angiokeratome das Untersuchungsergebnis mit zwei Bildern seines Patienten. Darauf kann man die, im Vergleich zur Aufnahme von 1898, fortschreitende Ausbreitung der Angiokeratome, speziell am Rücken und an den Armen erkennen. Im Urin konnte Fabry Albumin, das heißt eine Proteinurie, nachweisen. Diesem Befund schenkte er aber keine weitere Beachtung. Fabry stellte die Prognose auf, dass aufgrund des bisherigen Krankheitsverlaufes keine bedrohliche Entwicklung zu erwarten sei. Das Allgemeinbefinden sei nicht gestört und würde sich wohl auch in absehbarer Zeit nicht verändern. Eine Heilung erwartete er ebenfalls nicht, da die Hautveränderungen im Laufe der Jahre progressiv zunahmen. Von einer Behandlung sah Fabry ab, zumal sie auch von seinem Patienten nicht gewünscht war. Er hätte allerdings gerne einen Behandlungsversuch mit Röntgenstrahlung und Radium unternommen.
Fabry ging in dieser Veröffentlichung davon aus, dass sein Fall eines universellen Angiokeratoms in der Literatur kein Analogon habe: „Der einzige Fall, der in Frage käme, der Andersonsche, ist kein Angiokeratom, sondern es handelt sich um multiple Angiome.“[220] In diesem Punkt irrte sich Fabry.
1930, in seinem Todesjahr, veröffentlichte Fabry einen Artikel über das Angiokeratoma naeviforme. Darin ging er noch einmal auf den Fall seines Patienten ein. Emil Honke verstarb 1928 im Alter von 43 Jahren an einer nicht näher klassifizierten Lungenerkrankung, aber nicht an Tuberkulose.[217][221]
William Anderson und sein Patient


Im April 1898 publizierte der britische Anatom William Anderson im British Journal of Dermatology einen fünfseitigen Fallbericht über einen Patienten mit einem Angiokeratom (A case of ‘Angeio-keratoma’).[222]
William Anderson sah seinen Patienten (W. H.) erstmals im Dezember 1897. Dieser war zu diesem Zeitpunkt 39 Jahre alt und von Beruf Anstreicher. Bei ihm begannen die Hautveränderungen im Alter von elf Jahren; zunächst an den Knien, dann breiteten sie sich auf den Rumpf und die Extremitäten aus. Mit 17 war die maximale Ausbreitung erreicht.[217] Anderson hielt in einer Skizze, die Teil seiner Veröffentlichung war, die Ausbreitung der Angiokeratoma corporis diffusum fest. Bei der Familienanamnese stellten sich gleichartige Hautveränderungen bei der Mutter, der Schwester und drei von vier Kindern heraus. Bei der ersten Untersuchung fand Anderson etwas Albumin im Urin des Patienten. Danach war der Urin normal. Nach einigen Tagen im Krankenhaus nahmen die Läsionen etwas ab. Eine Behandlung erfolgte auf Wunsch des Patienten nicht.
Anderson ging davon aus, dass die Erkrankung bei beiden Geschlechtern vorkommt und von Frostbeulen beziehungsweise Asphyxie begleitet ist. Er äußerte sich zwar dahingehend, dass in vielen Fällen Familienmitglieder ebenfalls betroffen sein könnten, da aber in der Familie seines Patienten zum Zeitpunkt der Anamnese keine weiteren Fälle mit Angiokeratoma corporis diffusum bekannt waren, sah er keinen überzeugenden Beweis dafür, dass eine Erbkrankheit vorliegt.[217]
W. H. verstarb 1911 an einer Kachexie, die durch eine tuberkulöse Enteritis verursacht wurde. Eine Tochter und zwei Enkelkinder erkrankten ebenfalls an Morbus Fabry.[219][223]
Weitere Erforschung der Krankheit
In der Folgezeit wurde der Morbus Fabry zunächst als Angiokeratoma corporis diffusum bezeichnet. Die Bezeichnung Angiokeratom geht auf Ernest Wyndham Cottle (?–1919) zurück, der 1877 erstmals einen Fall von Angiokeratomen bei insgesamt acht Patienten mit ‚warzenartigen Geschwülsten‘ (warty growths) beschrieb.[224] Mit der Zeit bürgerte sich die heutige Bezeichnung Morbus Fabry ein, wobei Morbus Anderson-Fabry vor dem historischen Hintergrund angemessener wäre.[219]
Johannes Weicksel (1882–?) erkannte 1925 als erster die charakteristischen Veränderungen an der Retina und der Bindehaut als ein Symptom von Morbus Fabry.[225] Bei einem Brüderpaar wurde 1947 erstmals die Beteiligung der Nieren an der Progression von Morbus Fabry festgestellt. Bei der Autopsie entdeckte die Arbeitsgruppe um den Groninger Dermatologen Maximiliaan Ruiter (1900–1974) abnorme Vakuolen in den Blutgefäßen. Sie äußerten als erste den Verdacht, dass der Morbus Fabry eine systematisierte Speicherkrankheit sei.[226] In einigen Werken wird der Morbus Fabry daher auch als Ruiter-Pompen-Weyers-Syndrom bezeichnet.[227]
Der deutsche Pathologe Karl Scriba (1907–1978) vom Universitätsklinikum Hamburg-Eppendorf identifizierte in den Vakuolen im Jahr 1950 Lipide.[228] Drei Jahre später konnte er zusammen mit Hans Hornbostel die endothelialen Fettablagerungen erstmals bei einem lebenden Morbus-Fabry-Patienten nachweisen.[229]
1958 wurde erstmals ein Fall von Morbus Fabry bei einer Frau beschrieben.[199][230]
Charles C. Sweeley und Bernard Klionsky von der University of Pittsburgh charakterisierten 1963 die Fettablagerungen in den Nieren eines verstorbenen 28-jährigen Morbus-Fabry-Patienten und identifizierten dabei das Globotriaosylceramid. Sie klassifizierten die Krankheit als Sphingolipidose.[231]
Dass die Erkrankung einen X-chromosomal Erbgang aufweist, wurde 1965 von einer Arbeitsgruppe um John Opitz an der University of Wisconsin–Madison durch Stammbaumanalyse bewiesen.[232] Im selben Jahr entdeckte der in den Vereinigten Staaten lebende japanische Dermatologe Ken Hashimoto auf TEM-Aufnahmen von Hautproben von Morbus-Fabry-Patienten in den Zellen Einschlusskörperchen in den Lysosomen.[233] Er vermutete als erster, dass ein Defekt eines lysosomalen Enzyms die Ursache der Erkrankung ist.[234]
Entwicklung der Enzymersatztherapie
1967 wurde von einer Arbeitsgruppe am National Institute of Neurological Diseases and Blindness entdeckt, dass ein Defizit an Ceramidtrihexosidase die genetische Ursache von Morbus Fabry ist.[235] 1970 wurde das Enzym α-Galactosidase identifiziert,[236] von dem 1978 die beiden Varianten A und B aus menschlichen Plazenten isoliert werden konnte; ein Mangel an der Variante A ist für die Entstehung von Morbus Fabry verantwortlich.[237] Mit diesen Erkenntnissen konnten 1973 die Merkmalsträger – auch pränatal – auf biochemischem Weg anhand der Enzymaktivität bestimmt werden.[238]
Damit die Grundlage für die Enzymersatztherapie gelegt. Man begann zunächst mit der aufwändigen Isolierung des Enzyms aus humaner Plazenta,[239] Milz[240] sowie Plasma[241] und konnte nach der Infusion der α-Galactosidase A bei Morbus-Fabry-Patienten in deren Blut eine Reduzierung von Globotriaosylceramid feststellen.
Die Fortschritte der Gentechnik ermöglichten die Erzeugung von gentechnisch veränderten Organismen. Einer interdisziplinären Arbeitsgruppe unter dem Dach der National Institutes of Health gelang 1996 beim Modellorganismus Farbmaus der Knockout des α-Gal-A-Gens, wodurch die erste „Fabry-Maus“ geschaffen wurde.[242] Mit der Entwicklung der DNA-Sequenzierung konnten weitere Fortschritte in der Erforschung des Morbus Fabry erzielt werden. So ermöglichte die Kenntnis der Sequenz des GLA-Gens erstmals die Lokalisierung und Charakterisierung der Mutationen sowie die Herstellung rekombinanter DNA.[243][244][245]
Mit der in Bakterien eingeschleusten rekombinanten DNA konnte die α-Galactosidase A als rekombinantes Protein produziert und so in ausreichenden Mengen für präklinische und klinische Studien verwendet werden. Die ersten präklinischen Studien erfolgten 1996 an Fabry-Mäusen. Im Rahmen einer klinischen Studie erhielten 1999 erstmals Patienten die gentechnisch produzierte α-Galactosidase A.[246] Anfang August 2001 erfolgte in der Europäischen Union die zeitgleiche Zulassung von Agalsidase beta (Genzyme Corporation) und von Agalsidase alfa (Transkaryotic Therapies, jetzt Shire Pharmaceuticals), nach dem Orphan-Drug-Gesetz zur Behandlung des Morbus Fabry.[143][247] Am 15. August 2001 wurde Fabrazyme und Mitte Oktober desselben Jahres Replagal in Deutschland auf den Markt gebracht.[248]
Literatur
Fachbücher
- D. Elstein, G. Altarescu, M. Beck (Hrsg.): Fabry Disease. Verlag Springer, 2010, ISBN 978-90-481-9032-4 eingeschränkte Vorschau in der Google-Buchsuche
- J. A. Barranger, M. A. Cabrera-Salazar (Hrsg.): Lysosomal storage disorders. Verlag Springer, 2007, ISBN 978-0-387-70908-6, eingeschränkte Vorschau in der Google-Buchsuche
- C. Wanner: Morbus Fabry: Klinik, Diagnostik und Therapie. Verlag Uni-Med, 2004, ISBN 3-89599-769-2.
- A. Mehta, M. Beck, G. Sunder-Plassmann (Hrsg.): Fabry Disease. Oxford PharmaGenesis, 2006, ISBN 1-903539-03-X. PMID 21290683 (im Volltext frei zugänglich)
Fachartikel
- B. Hoffmann, E. Mayatepek: Morbus Fabry – oft gesehen, selten erkannt. In: Dtsch Arztebl Int. Band 106, 2009, S. 72–81. PMID 19623315
- B. Hoffmann: Fabry disease: recent advances in pathology, diagnosis, treatment and monitoring. In: Orphanet Journal of Rare Diseases. Band 4, 2009, S. 21. doi:10.1186/1750-1172-4-21, PMC 2768700 (freier Volltext). PMID 19818152 (Review-Artikel im Open Access)
- C. Whybra, M. Ries, C. Kampmann: Klinische Manifestation des Morbus Fabry bei Kindern. (PDF; 2,8 MB). In: Neuropädiatrie. Band 2, 2005, S. 44–48.
Sachbücher und populärwissenschaftliche Literatur
- Gerald Uhlig-Romero: Und trotzdem lebe ich. Mein Kampf mit einer rätselhaften Krankheit. Deutsche Verlags-Anstalt, 2009, ISBN 978-3-421-04326-9.
- U. Schmidt: Seltene Erbkrankheit. (PDF; 6,7 MB). In: JOGU. Band 186, 2003, S. 14–15.
- M. Pawlik: Gebt mir einen Ort ohne Angst! In: FAZ. 22. Juni 2009 (Rezension über Gerald Uhligs Buch)
- V. Hackenbroch: Fett im Organ. In: Der Spiegel. Nr. 28, 2006, S. 111 (online).
- N. Schäufler: Ein Waisenkind der Medizin – Die Erbkrankheit Morbus Fabry kennen selbst viele Ärzte nicht. In: FAZ. 12. Juni 2002, S. 10.
Weblinks
- Morbus Fabry. In: Online Mendelian Inheritance in Man. (englisch)
- Eintrag zu Fabry-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten)
- Morbus Fabry Pathologie – Bilddatenbank Pathopic der Universität Basel (PathoPic – Anleitung (PDF); PDF; 2,2 MB)
- Morbus Fabry Selbsthilfegruppe e. V.
- Stephanie Barbara Schatz: X-Inaktivierung bei heterozygoten Patientinnen bezüglich X-chromosomal vererbtem Morbus Fabry. (PDF; 9,1 MB) Dissertation, Ludwig-Maximilians-Universität München, 2012
Einzelnachweise
- ↑ B. J. Poorthuis, R. A. Wevers u. a.: The frequency of lysosomal storage diseases in The Netherlands. In: Human genetics. Band 105, Nummer 1–2, 1999 Jul-Aug, S. 151–156. PMID 10480370.
- ↑ P. J. Meikle, J. J. Hopwood u. a.: Prevalence of lysosomal storage disorders. In: JAMA. Band 281, Nummer 3, Januar 1999, S. 249–254, doi:10.1001/jama.281.3.249. PMID 9918480.
- ↑ a b R. J. Desnick, Y. A.Ioannou, C. M. Eng: a-Galactosidase A deficiency: Fabry disease. In: C. R. Scriver, A. L. Beaudet, W. S. Sly, D. Valle (Hrsg.): The metabolic and molecular basis of inherited disease. 8. Ausgabe, Verlag McGraw-Hill, 2001, ISBN 0-07-913035-6, S. 3733–3774.
- ↑ D. Marsden, H. Levy: Newborn screening of lysosomal storage disorders. In: Clinical chemistry. Band 56, Nummer 7, Juli 2010, S. 1071–1079, doi:10.1373/clinchem.2009.141622. PMID 20489136. (Review).
- ↑ M. Spada, S. Pagliardini u. a.: High incidence of later-onset fabry disease revealed by newborn screening. In: American journal of human genetics. Band 79, Nummer 1, Juli 2006, S. 31–40, doi:10.1086/504601. PMID 16773563. PMC 1474133 (freier Volltext).
- ↑ a b W. L. Hwu, Y. H. Chien u. a.: Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). In: Human mutation. Band 30, Nummer 10, Oktober 2009, S. 1397–1405, doi:10.1002/humu.21074. PMID 19621417. PMC 2769558 (freier Volltext).
- ↑ a b c d e f g h i j k l m n o p q r s t u v w x y z aa D. P. Germain: Fabry disease. In: Orphanet Journal of Rare Diseases. Band 5, 2010, S. 30, doi:10.1186/1750-1172-5-30. PMID 21092187. PMC 3009617 (freier Volltext). (Review im Open Access).
- ↑ Das Kompetenzzentrum für Morbus Fabry stellt sich vor. (PDF) (Seite nicht mehr abrufbar, festgestellt im März 2018. Suche in Webarchiven) (PDF; 595 kB) Uniklinik Köln, abgerufen am 1. September 2011
- ↑ a b C. Vetter: Repetitorium: Morbus Fabry. (PDF) In: ZM. Band 101, Nr. 1A, vom 1. Januar 2011, S. 41–45. (Seite nicht mehr abrufbar, festgestellt im März 2018. Suche in Webarchiven)
- ↑ a b K. F. Gold, G. M. Pastores u. a.: Quality of life of patients with Fabry disease. In: Qual Life Res. Band 11, Nummer 4, Juni 2002, S. 317–327. PMID 12086117.
- ↑ H. Y. Lin, K. W. Chong u. a.: High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. In: Circulation. Cardiovascular genetics. Band 2, Nummer 5, Oktober 2009, S. 450–456, doi:10.1161/CIRCGENETICS.109.862920. PMID 20031620.
- ↑ P. Gaspar, J. Herrera u. a.: Frequency of Fabry disease in male and female haemodialysis patients in Spain. In: BMC medical genetics. Band 11, 2010, S. 19, doi:10.1186/1471-2350-11-19. PMID 20122163. PMC 2837018 (freier Volltext).
- ↑ a b c d A. P. Burlina, K. B. Sims u. a.: Early diagnosis of peripheral nervous system involvement in Fabry disease and treatment of neuropathic pain: the report of an expert panel. In: BMC neurology. Band 11, 2011, S. 61, doi:10.1186/1471-2377-11-61. PMID 21619592. PMC 3126707 (freier Volltext).
- ↑ a b M. Pavelka, J. Roth: Funktionelle Ultrastruktur: Atlas der Biologie und Pathologie von Geweben. Verlag Springer, 2005, ISBN 3-211-83563-6, S. 104–105, doi:10.1007/3-211-30826-1_56 eingeschränkte Vorschau in der Google-Buchsuche
- ↑ omim.org: OMIM Gene/Loci: 413 – 422 of 718 on Chromosome X. Abgerufen am 1. September 2011
- ↑ M. W. King: Introduction to Fabry Disease. Stand: 13. Februar 2011, abgerufen am 1. September 2011
- ↑ K. Falke: Untersuchungen zur Mikrostruktur der Cornea verticillata bei Morbus Fabry und Vergleich zur medikamentös induzierten Form der Cornea verticillata mittels konfokaler In-vivo-Mikroskopie. Dissertation. Universität Rostock, 2009.
- ↑ H. F. Willard: The sex chromosomes and X chromosome inactivation. In: C. R. Scriver, A. L. Beaudet, W. S. Sly, D. Valle (Hrsg.): The metabolic and molecular basis of inherited disease. Band 3, 8. Ausgabe, MacGraw-Hill, 2001, ISBN 0-07-136322-X, S. 1191–1211.
- ↑ a b c L. L. Pinto, T. A. Vieira u. a.: Expression of the disease on female carriers of X-linked lysosomal disorders: a brief review. In: Orphanet Journal of Rare Diseases. Band 5, 2010, S. 14, doi:10.1186/1750-1172-5-14. PMID 20509947. PMC 2889886 (freier Volltext). (Review im Open Access)
- ↑ a b D. P. Germain, K. Benistan, L. Angelova: X-linked inheritance and its implication in the diagnosis and management of female patients in Fabry disease. In: La Revue de médecine interne. Band 31, Suppl 2, Dezember 2010, S. S209–S213, doi:10.1016/S0248-8663(10)70013-8. PMID 21211665.
- ↑ a b W. B. Dobyns, A. Filauro u. a.: Inheritance of most X-linked traits is not dominant or recessive, just X-linked. In: American journal of medical genetics. Part A. Band 129A, Nummer 2, August 2004, S. 136–143, doi:10.1002/ajmg.a.30123. PMID 15316978.
- ↑ W. B. Dobyns: The pattern of inheritance of X-linked traits is not dominant or recessive, just X-linked. In: Acta paediatrica. Band 95, Nummer 451, April 2006, S. 11–15, doi:10.1111/j.1651-2227.2006.tb02383.x. PMID 16720459.
- ↑ B. R. Migeon: X inactivation, female mosaicism, and sex differences in renal diseases. In: JASN. Band 19, Nummer 11, November 2008, S. 2052–2059, doi:10.1681/ASN.2008020198. PMID 18448583. (Review).
- ↑ M. Kobayashi, T. Ohashi u. a.: Clinical manifestations and natural history of Japanese heterozygous females with Fabry disease. In: Journal of inherited metabolic disease. [elektronische Veröffentlichung vor dem Druck] Januar 2008, doi:10.1007/s10545-007-0740-6. PMID 18202903.
- ↑ B. R. Migeon: Females Are Mosaics: X Inactivation and Sex Differences in Disease. Oxford University, New York 2007, ISBN 978-0-19-518812-7.
- ↑ I. Redonnet-Vernhet, J. K. Ploos van Amstel u. a.: Uneven X inactivation in a female monozygotic twin pair with Fabry disease and discordant expression of a novel mutation in the alpha-galactosidase A gene. In: Journal of medical genetics. Band 33, Nummer 8, August 1996, S. 682–688. PMID 8863162. PMC 1050704 (freier Volltext).
- ↑ E. M. Maier, S. Osterrieder u. a.: Disease manifestations and X inactivation in heterozygous females with Fabry disease. In: Acta paediatrica. Band 95, Nummer 451, April 2006, S. 30–38, doi:10.1111/j.1651-2227.2006.tb02386.x. PMID 16720462.
- ↑ a b c A. Gal, E. Schäfer, I. Rohard: The genetic basis of Fabry disease. In: A. Mehta, M. Beck, G. Sunder-Plassmann (Hrsg.): Fabry Disease: Perspectives from 5 Years of FOS. Kapitel 33, Oxford PharmaGenesis, 2006, ISBN 1-903539-03-X. PMID 21290673
- ↑ A. Gal: Molecular genetics of Fabry disease and Genotype-phenotype correlation. In: D. Elstein, G. Altarescu, M. Beck (Hrsg.): Fabry disease. Verlag Springer, 2010, ISBN 978-90-481-9032-4, S. 3–19. eingeschränkte Vorschau in der Google-Buchsuche
- ↑ R. O. Brady: Fabry Disease – An Overview. In: D. Elstein, G. Altarescu, M. Beck (Hrsg.): Fabry Disease. Verlag Springer, 2010, ISBN 978-90-481-9032-4, S. XIX eingeschränkte Vorschau in der Google-Buchsuche
- ↑ a b M. D. Sanchez-Niño, A. B. Sanz u. a.: Globotriaosylsphingosine actions on human glomerular podocytes: implications for Fabry nephropathy. In: Nephrology, dialysis, transplantation. Band 26, Nummer 6, Juni 2011, S. 1797–1802, doi:10.1093/ndt/gfq306. PMID 20504837.
- ↑ a b A. H. Futerman, G. van Meer: The cell biology of lysosomal storage disorders. In: Nature Reviews Molecular Cell Biology. Band 5, Nummer 7, Juli 2004, S. 554–565, doi:10.1038/nrm1423. PMID 15232573. (Review).
- ↑ M. Fuller: Sphingolipids: the nexus between Gaucher disease and insulin resistance. In: Lipids in health and disease. Band 9, 2010, S. 113, doi:10.1186/1476-511X-9-113. PMID 20937139. PMC 2964722 (freier Volltext). (Review im Open Access).
- ↑ a b c A. C. Vedder, G. E. Linthorst u. a.: Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. In: PloS one. Band 2, Nummer 7, 2007, S. e598, doi:10.1371/journal.pone.0000598. PMID 17622343. PMC 1913555 (freier Volltext). (Open Access)
- ↑ K. D. MacDermot, A. Holmes, A. H. Miners: Natural history of Fabry disease in affected males and obligate carrier females. In: Journal of inherited metabolic disease. Band 24 Suppl 2, 2001, S. 13–14. PMID 11758673.
- ↑ C. Whybra, C. Kampmann u. a.: Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. In: Journal of inherited metabolic disease. Band 24, Nummer 7, Dezember 2001, S. 715–724. PMID 11804208.
- ↑ P. B. Deegan, A. F. Baehner u. a.: Natural history of Fabry disease in females in the Fabry Outcome Survey. In: Journal of medical genetics. Band 43, Nummer 4, April 2006, S. 347–352, doi:10.1136/jmg.2005.036327. PMID 16227523. PMC 2563231 (freier Volltext).
- ↑ S. Gupta, M. Ries u. a.: The relationship of vascular glycolipid storage to clinical manifestations of Fabry disease: a cross-sectional study of a large cohort of clinically affected heterozygous women. In: Medicine. Band 84, Nummer 5, September 2005, S. 261–268. PMID 16148726.
- ↑ a b c A. C. Vedder, G. E. Linthorst u. a.: The Dutch Fabry cohort: diversity of clinical manifestations and Gb3 levels. In: Journal of inherited metabolic disease. Band 30, Nummer 1, Februar 2007, S. 68–78, doi:10.1007/s10545-006-0484-8. PMID 17206462.
- ↑ A. C. Vedder, A. Strijland u. a.: Manifestations of Fabry disease in placental tissue. In: Journal of inherited metabolic disease. Band 29, Nummer 1, Februar 2006, S. 106–111, doi:10.1007/s10545-006-0196-0. PMID 16601876.
- ↑ a b E. Young, K. Mills u. a.: Is globotriaosylceramide a useful biomarker in Fabry disease? In: Acta paediatrica. Band 94, Nummer 447, März 2005, S. 51–54. PMID 15895713.
- ↑ S. Bekri, O. Lidove u. a.: The role of ceramide trihexoside (globotriaosylceramide) in the diagnosis and follow-up of the efficacy of treatment of Fabry disease: a review of the literature. In: Cardiovascular & hematological agents in medicinal chemistry. Band 4, Nummer 4, Oktober 2006, S. 289–297. PMID 17073606. (Review).
- ↑ J. M. Aerts, J. E. Groener u. a.: Elevated globotriaosylsphingosine is a hallmark of Fabry disease. In: PNAS. Band 105, Nummer 8, Februar 2008, S. 2812–2817, doi:10.1073/pnas.0712309105. PMID 18287059. PMC 2268542 (freier Volltext).
- ↑ a b A. H. Miners, A. Holmes u. a.: Assessment of health-related quality-of-life in males with Anderson Fabry Disease before therapeutic intervention. In: Quality of life research. Band 11, Nummer 2, März 2002, S. 127–133. PMID 12018736.
- ↑ a b D. A. Laney, D. J. Gruskin u. a.: Social-adaptive and psychological functioning of patients affected by Fabry disease. In: Journal of inherited metabolic disease. Januar 2010, doi:10.1007/s10545-009-9025-6. PMID 20087663.
- ↑ a b A. J. Grau, M. Schwaninger u. a.: Eine lysosomale Stoffwechselerkrankung mit neuen therapeutischen Möglichkeiten. In: Der Nervenarzt. Band 74, Nummer 6, Juni 2003, S. 489–496, doi:10.1007/s00115-003-1513-6. PMID 12799787.
- ↑ J. Sadek, R. Shellhaas u. a.: Psychiatric findings in four female carriers of Fabry disease. In: Psychiatric genetics. Band 14, Nummer 4, Dezember 2004, S. 199–201. PMID 15564893.
- ↑ a b A. L. Cole, P. J. Lee u. a.: Depression in adults with Fabry disease: a common and under-diagnosed problem. In: Journal of inherited metabolic disease. Band 30, Nummer 6, November 2007, S. 943–951, doi:10.1007/s10545-007-0708-6. PMID 17994284.
- ↑ P. Segal, Y. Kohn u. a.: Psychiatric and cognitive profile in Anderson-Fabry patients: a preliminary study. In: Journal of inherited metabolic disease. Band 33, Nummer 4, August 2010, S. 429–436, doi:10.1007/s10545-010-9133-3. PMID 20549363.
- ↑ T. W. Crosbie, W. Packman, S. Packman: Psychological aspects of patients with Fabry disease. In: Journal of inherited metabolic disease. Band 32, Nummer 6, Dezember 2009, S. 745–753, doi:10.1007/s10545-009-1254-1. PMID 19924564.
- ↑ W. J. Cable, E. H. Kolodny, R. D. Adams: Fabry disease: impaired autonomic function. In: Neurology. Band 32, Nummer 5, Mai 1982, S. 498–502. PMID 6803189.
- ↑ M. Dütsch, H. Marthol u. a.: Small fiber dysfunction predominates in Fabry neuropathy. In: Journal of clinical neurophysiology. Band 19, Nummer 6, Dezember 2002, S. 575–586. PMID 12488789.
- ↑ B. Hoffmann, M. Beck u. a.: Nature and prevalence of pain in Fabry disease and its response to enzyme replacement therapy–a retrospective analysis from the Fabry Outcome Survey. In: The Clinical journal of pain. Band 23, Nummer 6, Jul-Aug 2007, S. 535–542, doi:10.1097/AJP.0b013e318074c986. PMID 17575495.
- ↑ a b R. J. Hopkin, J. Bissler u. a.: Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. In: Pediatric research. Band 64, Nummer 5, November 2008, S. 550–555, doi:10.1203/PDR.0b013e318183f132. PMID 18596579.
- ↑ J. Charrow: A 14-year-old boy with pain in hands and feet. In: Pediatric annals. Band 38, Nummer 4, April 2009, S. 190, 192. PMID 19455947.
- ↑ M. J. Hilz, B. Stemper, E. H. Kolodny: Lower limb cold exposure induces pain and prolonged small fiber dysfunction in Fabry patients. In: Pain. Band 84, Nummer 2–3, Februar 2000, S. 361–365. PMID 10666542.
- ↑ K. J. Sheth, S. L. Werlin u. a.: Gastrointestinal structure and function in Fabry’s disease. In: The American Journal of Gastroenterology. Band 76, Nummer 3, September 1981, S. 246–251. PMID 6274188.
- ↑ a b B. Hoffmann, M. Schwarz u. a.: Gastrointestinal symptoms in 342 patients with Fabry disease: prevalence and response to enzyme replacement therapy. In: Clinical gastroenterology and hepatology. Band 5, Nummer 12, Dezember 2007, S. 1447–1453, doi:10.1016/j.cgh.2007.08.012. PMID 17919989.
- ↑ W. H. Kang, S. I. Chun, S. Lee: Generalized anhidrosis associated with Fabry’s disease. In: Journal of the American Academy of Dermatology. Band 17, Nummer 5 Pt 2, November 1987, S. 883–887. PMID 3119682.
- ↑ a b c d C. H. Orteu, T. Jansen u. a.: Fabry disease and the skin: data from FOS, the Fabry outcome survey. In: The British journal of dermatology. Band 157, Nummer 2, August 2007, S. 331–337, doi:10.1111/j.1365-2133.2007.08002.x. PMID 17573884.
- ↑ a b S. N. Gupta, M. Ries u. a.: Skin-impedance in Fabry Disease: a prospective, controlled, non-randomized clinical study. In: BMC neurology. Band 8, 2008, S. 41, doi:10.1186/1471-2377-8-41. PMID 18990229. PMC 2585087 (freier Volltext). (Open Access)
- ↑ a b E. D. Shelley, W. B. Shelley, T. W. Kurczynski: Painful fingers, heat intolerance, and telangiectases of the ear: easily ignored childhood signs of Fabry disease. In: Pediatric dermatology. Band 12, Nummer 3, September 1995, S. 215–219. PMID 7501549.
- ↑ C. M. Eng, D. P. Germain u. a.: Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. In: Genetics in medicine. Band 8, Nummer 9, September 2006, S. 539–548, doi:10.1097/01.gim.0000237866.70357.c6. PMID 16980809. (Review).
- ↑ M. Möhrenschlager, M. Braun-Falco u. a.: Fabry disease: recognition and management of cutaneous manifestations. In: American journal of clinical dermatology. Band 4, Nummer 3, 2003, S. 189–196. PMID 12627994. (Review).
- ↑ D. Wattanasirichaigoon, J. Svasti u. a.: Clinical and molecular characterization of an extended family with Fabry disease. In: Journal of the Medical Association of Thailand. Band 89, Nummer 9, September 2006, S. 1528–1535. PMID 17100396.
- ↑ C. Orssaud, J. Dufier, D. Germain: Ocular manifestations in Fabry disease: a survey of 32 hemizygous male patients. In: Ophthalmic genetics. Band 24, Nummer 3, September 2003, S. 129–139. PMID 12868031.
- ↑ T. T. Nguyen, T. Gin u. a.: Ophthalmological manifestations of Fabry disease: a survey of patients at the Royal Melbourne Fabry Disease Treatment Centre. In: Clinical & experimental ophthalmology. Band 33, Nummer 2, April 2005, S. 164–168, doi:10.1111/j.1442-9071.2005.00990.x. PMID 15807825.
- ↑ A. Sodi, A. S. Ioannidis u. a.: Ocular manifestations of Fabry’s disease: data from the Fabry Outcome Survey. In: The British journal of ophthalmology. Band 91, Nummer 2, Februar 2007, S. 210–214, doi:10.1136/bjo.2006.100602. PMID 16973664. PMC 1857640 (freier Volltext).
- ↑ K. Falke, A. Büttner u. a.: The microstructure of cornea verticillata in Fabry disease and amiodarone-induced keratopathy: a confocal laser-scanning microscopy study. In: Graefe’s archive for clinical and experimental ophthalmology. Band 247, Nummer 4, April 2009, S. 523–534, doi:10.1007/s00417-008-0962-9. PMID 18931853.
- ↑ A. Sessa, M. Meroni u. a.: Renal pathological changes in Fabry disease. In: Journal of inherited metabolic disease. Band 24 Suppl 2, 2001, S. 66–70. PMID 11758681. (Review).
- ↑ A. Sessa, M. Meroni u. a.: Evolution of renal pathology in Fabry disease. In: Acta paediatrica. Band 92, Nummer 443, Dezember 2003, S. 6–8. PMID 14989458.
- ↑ a b A. B. Fogo, L. Bostad u. a.: Scoring system for renal pathology in Fabry disease: report of the International Study Group of Fabry Nephropathy (ISGFN). In: Nephrology, dialysis, transplantation. Band 25, Nummer 7, Juli 2010, S. 2168–2177, doi:10.1093/ndt/gfp528. PMID 19833663. PMC 2902894 (freier Volltext).
- ↑ M. C. Gubler, G. Lenoir u. a.: Early renal changes in hemizygous and heterozygous patients with Fabry’s disease. In: Kidney International. Band 13, Nummer 3, März 1978, S. 223–235. PMID 418264.
- ↑ J. Alroy, S. Sabnis, J. B. Kopp: Renal pathology in Fabry disease. In: JASN. Band 13 Suppl 2, Juni 2002, S. S134–S138. PMID 12068025. (Review).
- ↑ a b c d e T. Thomaidis, M. Relle u. a.: Wirkung der Enzymersatztherapie (ERT) auf die Nierenfunktion von Patienten mit Morbus Fabry. In: Medizinische Klinik. Band 104, Nummer 9, September 2009, S. 699–703, doi:10.1007/s00063-009-1152-1. PMID 19779674. (Review).
- ↑ A. Ortiz, J. P. Oliveira u. a.: Nephropathy in males and females with Fabry disease: cross-sectional description of patients before treatment with enzyme replacement therapy. In: Nephrology, dialysis, transplantation. Band 23, Nummer 5, Mai 2008, S. 1600–1607, doi:10.1093/ndt/gfm848. PMID 18175781.
- ↑ a b M. H. Branton, R. Schiffmann u. a.: Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. In: Medicine. Band 81, Nummer 2, März 2002, S. 122–138. PMID 11889412.
- ↑ a b c d R. Schiffmann, D. G. Warnock u. a.: Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. In: Nephrology, dialysis, transplantation. Band 24, Nummer 7, Juli 2009, S. 2102–2111, doi:10.1093/ndt/gfp031. PMID 19218538. PMC 2698092 (freier Volltext).
- ↑ R. B. Glass u. a.: Fabry disease: renal sonographic and magnetic resonance imaging findings in affected males and carrier females with the classic and cardiac variant phenotypes. In: J Comput Assist Tomogr. Band 28, 2004, S. 158–168.
- ↑ a b c A. Linhart, T. Palecek u. a.: New insights in cardiac structural changes in patients with Fabry’s disease. In: American Heart Journal. Band 139, Nummer 6, Juni 2000, S. 1101–1108, doi:10.1067/mhj.2000.105105. PMID 10827394.
- ↑ a b c C. Kampmann, F. Baehner u. a.: Cardiac manifestations of Anderson-Fabry disease in heterozygous females. In: Journal of the American College of Cardiology. Band 40, Nummer 9, November 2002, S. 1668–1674. PMID 12427421.
- ↑ a b M. Senechal, D. P. Germain: Fabry disease: a functional and anatomical study of cardiac manifestations in 20 hemizygous male patients. In: Clinical genetics. Band 63, Nummer 1, Januar 2003, S. 46–52. PMID 12519371.
- ↑ a b J. S. Shah, D. A. Hughes u. a.: Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. In: The American journal of cardiology. Band 96, Nummer 6, September 2005, S. 842–846, doi:10.1016/j.amjcard.2005.05.033. PMID 16169374.
- ↑ P. M. Elliott, H. Kindler u. a.: Coronary microvascular dysfunction in male patients with Anderson-Fabry disease and the effect of treatment with alpha galactosidase A. In: Heart. Band 92, Nummer 3, März 2006, S. 357–360, doi:10.1136/hrt.2004.054015. PMID 16085718. PMC 1860797 (freier Volltext).
- ↑ J. C. Moon, M. Sheppard u. a.: The histological basis of late gadolinium enhancement cardiovascular magnetic resonance in a patient with Anderson-Fabry disease. In: J Cardiovasc Magn Reson. Band 8, Nummer 3, 2006, S. 479–482. PMID 16755835.
- ↑ H. Hasegawa, H. Takano u. a.: Images in cardiovascular medicine. Transition from left ventricular hypertrophy to massive fibrosis in the cardiac variant of Fabry disease. In: Circulation. Band 113, Nummer 16, April 2006, S. e720–e721, doi:10.1161/CIRCULATIONAHA.105.584292. PMID 16636179.
- ↑ F. Weidemann, F. Breunig u. a.: The variation of morphological and functional cardiac manifestation in Fabry disease: potential implications for the time course of the disease. In: European heart journal. Band 26, Nummer 12, Juni 2005, S. 1221–1227, doi:10.1093/eurheartj/ehi143. PMID 15728649.
- ↑ A. Linhart, C. Kampmann u. a.: Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. In: European heart journal. Band 28, Nummer 10, Mai 2007, S. 1228–1235, doi:10.1093/eurheartj/ehm153. PMID 17483538.
- ↑ C. Kampmann, A. Linhart u. a.: Onset and progression of the Anderson-Fabry disease related cardiomyopathy. In: International journal of cardiology. Band 130, Nummer 3, November 2008, S. 367–373, doi:10.1016/j.ijcard.2008.03.007. PMID 18572264.
- ↑ a b T. Takenaka, H. Teraguchi u. a.: Terminal stage cardiac findings in patients with cardiac Fabry disease: an electrocardiographic, echocardiographic, and autopsy study. In: Journal of cardiology. Band 51, Nummer 1, Februar 2008, S. 50–59, doi:10.1016/j.jjcc.2007.12.001. PMID 18522775.
- ↑ a b M. Pieroni, C. Chimenti u. a.: Early detection of Fabry cardiomyopathy by tissue Doppler imaging. In: Circulation. Band 107, Nummer 15, April 2003, S. 1978–1984, doi:10.1161/01.CIR.0000061952.27445.A0. PMID 12668521.
- ↑ F. Weidemann, F. Breunig u. a.: Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease: a prospective strain rate imaging study. In: Circulation. Band 108, Nummer 11, September 2003, S. 1299–1301, doi:10.1161/01.CIR.0000091253.71282.04. PMID 12952834.
- ↑ M. Pieroni, C. Chimenti u. a.: Tissue Doppler imaging in Fabry disease. In: Current opinion in cardiology. Band 19, Nummer 5, September 2004, S. 452–457. PMID 15316452. (Review).
- ↑ T. Palecek, G. Dostalova u. a.: Right ventricular involvement in Fabry disease. In: Journal of the American Society of Echocardiography. Band 21, Nummer 11, November 2008, S. 1265–1268, doi:10.1016/j.echo.2008.09.002. PMID 18835697.
- ↑ M. Niemann, F. Breunig u. a.: The right ventricle in Fabry disease: natural history and impact of enzyme replacement therapy. In: Heart. Band 96, Nummer 23, Dezember 2010, S. 1915–1919, doi:10.1136/hrt.2010.204586. PMID 20965976.
- ↑ C. Kampmann, F. A. Baehner u. a.: The right ventricle in Fabry disease. In: Acta paediatrica. Band 94, Nummer 447, März 2005, S. 15–18. PMID 15895706.
- ↑ a b A. Fellgiebel, M. J. Müller, L. Ginsberg: CNS manifestations of Fabry’s disease. In: Lancet neurology. Band 5, Nummer 9, September 2006, S. 791–795, doi:10.1016/S1474-4422(06)70548-8. PMID 16914407.
- ↑ a b c K. Sims, J. Politei u. a.: Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. In: Stroke. Band 40, Nummer 3, März 2009, S. 788–794, doi:10.1161/STROKEAHA.108.526293. PMID 19150871.
- ↑ a b P. Mitsias, S. R. Levine: Cerebrovascular complications of Fabry’s disease. In: Annals of neurology. Band 40, Nummer 1, Juli 1996, S. 8–17, doi:10.1002/ana.410400105. PMID 8687196.
- ↑ M. F. Mendez, T. M. Stanley u. a.: The vascular dementia of Fabry’s disease. In: Dementia and geriatric cognitive disorders. Band 8, Nummer 4, 1997 Jul-Aug, S. 252–257. PMID 9213072.
- ↑ R. Okeda, M. Nisihara: An autopsy case of Fabry disease with neuropathological investigation of the pathogenesis of associated dementia. In: Neuropathology. Band 28, Nummer 5, Oktober 2008, S. 532–540, doi:10.1111/j.1440-1789.2008.00883.x. PMID 18410273.
- ↑ A. Fellgiebel, I. Keller u. a.: Diagnostic utility of different MRI and MR angiography measures in Fabry disease. In: Neurology. Band 72, Nummer 1, Januar 2009, S. 63–68, doi:10.1212/01.wnl.0000338566.54190.8a. PMID 19122032.
- ↑ T. DeGraba, S. Azhar u. a.: Profile of endothelial and leukocyte activation in Fabry patients. In: Annals of neurology. Band 47, Nummer 2, Februar 2000, S. 229–233. PMID 10665494.
- ↑ D. F. Moore, L. T. Scott u. a.: Regional cerebral hyperperfusion and nitric oxide pathway dysregulation in Fabry disease: reversal by enzyme replacement therapy. In: Circulation. Band 104, Nummer 13, September 2001, S. 1506–1512. PMID 11571244.
- ↑ D. F. Moore, G. Altarescu u. a.: Elevated cerebral blood flow velocities in Fabry disease with reversal after enzyme replacement. In: Stroke; a journal of cerebral circulation. Band 33, Nummer 2, Februar 2002, S. 525–531. PMID 11823664.
- ↑ R. Schiffmann: Fabry disease. In: Pharmacology & Therapeutics. Band 122, Nummer 1, April 2009, S. 65–77, doi:10.1016/j.pharmthera.2009.01.003. PMID 19318041. (Review).
- ↑ C. R. Kaneski, D. F. Moore u. a.: Myeloperoxidase predicts risk of vasculopathic events in hemizgygous males with Fabry disease. In: Neurology. Band 67, Nummer 11, Dezember 2006, S. 2045–2047, doi:10.1212/01.wnl.0000247278.88077.09. PMID 17159117. PMC 1950664 (freier Volltext).
- ↑ A. Palla, S. Hegemann u. a.: Vestibular and auditory deficits in Fabry disease and their response to enzyme replacement therapy. In: Journal of neurology. Band 254, Nummer 10, Oktober 2007, S. 1433–1442, doi:10.1007/s00415-007-0575-y. PMID 17934877.
- ↑ Y. Sakurai, H. Kojima u. a.: The hearing status in 12 female and 15 male Japanese Fabry patients. In: Auris, nasus, larynx. Band 36, Nummer 6, Dezember 2009, S. 627–632, doi:10.1016/j.anl.2009.01.001. PMID 19261412.
- ↑ D. P. Germain, P. Avan u. a.: Patients affected with Fabry disease have an increased incidence of progressive hearing loss and sudden deafness: an investigation of twenty-two hemizygous male patients. In: BMC medical genetics. Band 3, Oktober 2002, S. 10. PMID 12377100. PMC 134464 (freier Volltext).
- ↑ G. Conti, B. Sergi: Auditory and vestibular findings in Fabry disease: a study of hemizygous males and heterozygous females. In: Acta paediatrica. Band 92, Nummer 443, Dezember 2003, S. 33–37. PMID 14989464.
- ↑ D. M. Rosenberg, V. J. Ferrans u. a.: Chronic airflow obstruction in Fabry’s disease. In: The American journal of medicine. Band 68, Nummer 6, Juni 1980, S. 898–905. PMID 6247911.
- ↑ L. K. Brown, A. Miller u. a.: Pulmonary involvement in Fabry disease. In: American journal of respiratory and critical care medicine. Band 155, Nummer 3, März 1997, S. 1004–1010. PMID 9116979.
- ↑ S. Magage, J. C. Lubanda u. a.: Atteinte respiratoire de la maladie de Fabry. In: Médecine sciences. Band 21, Nummer 11 Suppl, Dezember 2005, S. 37–39, doi:10.1051/medsci/20052111s37. PMID 16324666.
- ↑ S. Magage, J. C. Lubanda u. a.: Natural history of the respiratory involvement in Anderson-Fabry disease. In: Journal of inherited metabolic disease. Band 30, Nummer 5, Oktober 2007, S. 790–799, doi:10.1007/s10545-007-0616-9. PMID 17619837.
- ↑ D. P. Germain, K. Benistan u. a.: Atteinte osseuse de la maladie de Fabry. In: Médecine sciences. Band 21, Nummer 11 Suppl, Dezember 2005, S. 43–44, doi:10.1051/medsci/20052111s43. PMID 16324668.
- ↑ D. P. Germain, K. Benistan u. a.: Osteopenia and osteoporosis: previously unrecognized manifestations of Fabry disease. In: Clinical genetics. Band 68, Nummer 1, Juli 2005, S. 93–95, doi:10.1111/j.1399-0004.2005.00457.x. PMID 15952993.
- ↑ H. Mersebach, J. O. Johansson u. a.: Osteopenia: a common aspect of Fabry disease. Predictors of bone mineral density. In: Genetics in medicine. Band 9, Nummer 12, Dezember 2007, S. 812–818, doi:10.1097/GIM.0b013e31815cb197. PMID 18091430.
- ↑ R. O. Brady: Bone and muscle involvement in Fabry disease. In: D. Elstein, G. Altarescu, M. Beck (Hrsg.): Fabry Disease. Verlag Springer, 2010, ISBN 978-90-481-9032-4, S. 293–298 eingeschränkte Vorschau in der Google-Buchsuche
- ↑ C. L. Marchesoni, N. Roa u. a.: Misdiagnosis in Fabry disease. In: The Journal of pediatrics. Band 156, Nummer 5, Mai 2010, S. 828–831, doi:10.1016/j.jpeds.2010.02.012. PMID 20385321.
- ↑ A. Mehta, R. Ricci u. a.: Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. In: European journal of clinical investigation. Band 34, Nummer 3, März 2004, S. 236–242, doi:10.1111/j.1365-2362.2004.01309.x. PMID 15025684.
- ↑ a b c K. D. MacDermot, A. Holmes, A. H. Miners: Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. In: Journal of medical genetics. Band 38, Nummer 11, November 2001, S. 750–760. PMID 11694547. PMC 173476 (freier Volltext).
- ↑ N. Siegmund-Schultze: Morbus Fabry ist selten, die Symptomatik unspezifisch – Vor der Diagnose steht oft eine Odyssee von Arzt zu Arzt. In: Ärzte Zeitung. 15. Dezember 2004
- ↑ J. S. Mayes, J. B. Scheerer u. a.: Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry’s disease. In: Clinica chimica acta. Band 112, Nummer 2, Mai 1981, S. 247–251. PMID 6263521.
- ↑ B. Hoffmann, H. Georg Koch u. a.: Deficient alpha-galactosidase A activity in plasma but no Fabry disease–a pitfall in diagnosis. In: Clinical chemistry and laboratory medicine. Band 43, Nummer 11, 2005, S. 1276–1277, doi:10.1515/CCLM.2005.219. PMID 16232095.
- ↑ G. E. Linthorst, A. C. Vedder u. a.: Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers. In: Clinica chimica acta. Band 353, Nummer 1–2, März 2005, S. 201–203, doi:10.1016/j.cccn.2004.10.019. PMID 15698608.
- ↑ a b B. E. Smid, L. van der Tol, M. Biegstraaten, G. E. Linthorst, C. E. M. Hollak, B. J. H. M. Poorthuis: Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. In: J Med Genet. Band 52, Nr. 4, 2015, S. 262–226.
- ↑ S. Roy, K. Gaudin u. a.: Optimisation of the separation of four major neutral glycosphingolipids: application to a rapid and simple detection of urinary globotriaosylceramide in Fabry disease. In: Journal of chromatography B. Band 805, Nummer 2, Juni 2004, S. 331–337, doi:10.1016/j.jchromb.2004.03.037. PMID 15135109.
- ↑ C. Auray-Blais, D. Cyr u. a.: Development of a filter paper method potentially applicable to mass and high-risk urinary screenings for Fabry disease. In: Journal of inherited metabolic disease. Band 30, Nummer 1, Februar 2007, S. 106, doi:10.1007/s10545-006-0444-3. PMID 17171433.
- ↑ C. Auray-Blais, D. Cyr u. a.: Urinary globotriaosylceramide excretion correlates with the genotype in children and adults with Fabry disease. In: Molecular Genetics and Metabolism. Band 93, Nummer 3, März 2008, S. 331–340, doi:10.1016/j.ymgme.2007.10.001. PMID 18023222.
- ↑ D. Touboul, S. Roy u. a.: Fast fingerprinting by MALDI-TOF mass spectrometry of urinary sediment glycosphingolipids in Fabry disease. In: Analytical and bioanalytical chemistry. Band 382, Nummer 5, Juli 2005, S. 1209–1216, doi:10.1007/s00216-005-3239-8. PMID 15959771.
- ↑ K. Mills, P. Morris u. a.: Measurement of urinary CDH and CTH by tandem mass spectrometry in patients hemizygous and heterozygous for Fabry disease. In: Journal of inherited metabolic disease. Band 28, Nummer 1, 2005, S. 35–48, doi:10.1007/s10545-005-5263-4. PMID 15702404.
- ↑ R. J. Desnick: Prenatal diagnosis of Fabry disease. In: Prenatal diagnosis. Band 27, Nummer 8, August 2007, S. 693–694, doi:10.1002/pd.1767. PMID 17533632. (Review).
- ↑ C. Lindengrün: Seltene Leiden – Gesichter der Mukopolysaccharidosen. In: CliniCum. 9, 2010.
- ↑ A. M. Das: Angeborene Stoffwechselstörungen. In: A. Rieder, B. Lohff (Hrsg.): Gender Medizin. Verlag Springer, 2004, ISBN 3-211-00766-0, S. 67f.eingeschränkte Vorschau in der Google-Buchsuche
- ↑ T. F. Metz, T. P. Mechtler u. a.: Simplified newborn screening protocol for lysosomal storage disorders. In: Clinical chemistry. Band 57, Nummer 9, September 2011, S. 1286–1294, doi:10.1373/clinchem.2011.164640. PMID 21771947.
- ↑ K. Nakamura, K. Hattori, F. Endo: Newborn screening for lysosomal storage disorders. In: American journal of medical genetics. Band 157, Nummer 1, Februar 2011, S. 63–71, doi:10.1002/ajmg.c.30291. PMID 21312327. (Review).
- ↑ M. Spada, D. Kasper, V. Pagliardini, E. Biamino, S. Giachero, F. Porta: Metabolic progression to clinical phenotype in classic Fabry disease. In: Ital J Pediatr. Band 43, Nr. 1, 2017, S. 1.
- ↑ G. M. Pastores: Miglustat: Substrate Reduction Therapy for Lysosomal Storage Disorders Associated with Primary Central Nervous System Involvement. ( vom 16. Juni 2010 im Internet Archive) (PDF; 75 kB). In: Recent Patents on CNS Drug Discovery. 1, 2006, S. 77–82. PMID 18221193
- ↑ a b Therapie des Morbus Fabry. ( vom 3. Februar 2015 im Internet Archive) Abgerufen am 13. September 2011
- ↑ J. M. Donnelly: Shire wins FDA approval for rare disease drug. In: Boston Business Journal. 25. August 2011
- ↑ a b C. A. Black: March on, not in. In: Nature medicine. Band 17, Nummer 5, Mai 2011, S. 515, doi:10.1038/nm0511-515. PMID 21546944.
- ↑ a b M. Beck: Agalsidase alfa – a preparation for enzyme replacement therapy in Anderson-Fabry disease. In: Expert opinion on investigational drugs. Band 11, Nummer 6, Juni 2002, S. 851–858, doi:10.1517/13543784.11.6.851. PMID 12036428. (Review).
- ↑ K. Lee, X. Jin u. a.: A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. In: Glycobiology. Band 13, Nummer 4, April 2003, S. 305–313, doi:10.1093/glycob/cwg034. PMID 12626384.
- ↑ Fachinformation Fabrazyme 5mg, 35mg, Stand 11/2018
- ↑ a b W. R. Wilcox, M. Banikazemi u. a.: Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. In: American journal of human genetics. Band 75, Nummer 1, Juli 2004, S. 65–74, doi:10.1086/422366. PMID 15154115. PMC 1182009 (freier Volltext).
- ↑ B. L. Thurberg, H. Rennke u. a.: Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. In: Kidney International. Band 62, Nummer 6, Dezember 2002, S. 1933–1946, doi:10.1046/j.1523-1755.2002.00675.x. PMID 12427118.
- ↑ a b c d J. Marshall, K. M. Ashe u. a.: Substrate reduction augments the efficacy of enzyme therapy in a mouse model of Fabry disease. In: PloS one. Band 5, Nummer 11, 2010, S. e15033, doi:10.1371/journal.pone.0015033. PMID 21124789. PMC 2991350 (freier Volltext). (Open Access)
- ↑ M. Beck: Agalsidase alfa for the treatment of Fabry disease: new data on clinical efficacy and safety. In: Expert opinion on biological therapy. Band 9, Nummer 2, Februar 2009, S. 255–261, doi:10.1517/14712590802658428. PMID 19236256. (Review).
- ↑ a b C. M. Eng, N. Guffon u. a.: Safety and efficacy of recombinant human alpha-galactosidase A–replacement therapy in Fabry’s disease. In: The New England Journal of Medicine. Band 345, Nummer 1, Juli 2001, S. 9–16, doi:10.1056/NEJM200107053450102. PMID 11439963.
- ↑ G. M. Keating, D. Simpson: Spotlight on agalsidase beta in Fabry disease. In: BioDrugs. Band 21, Nummer 4, 2007, S. 269–271. PMID 17628124. (Review).
- ↑ D. P. Germain, S. Waldek u. a.: Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. In: JASN. Band 18, Nummer 5, Mai 2007, S. 1547–1557, doi:10.1681/ASN.2006080816. PMID 17409312.
- ↑ a b Y. Tajima, I. Kawashima u. a.: Use of a modified alpha-N-acetylgalactosaminidase in the development of enzyme replacement therapy for Fabry disease. In: American journal of human genetics. Band 85, Nummer 5, November 2009, S. 569–580, doi:10.1016/j.ajhg.2009.09.016. PMID 19853240. PMC 2775840 (freier Volltext).
- ↑ T. Ohashi, M. Sakuma u. a.: Influence of antibody formation on reduction of globotriaosylceramide (GL-3) in urine from Fabry patients during agalsidase beta therapy. In: Molecular Genetics and Metabolism. Band 92, Nummer 3, November 2007, S. 271–273, doi:10.1016/j.ymgme.2007.06.013. PMID 17689998.
- ↑ T. Ohashi, S. Iizuka u. a.: Reduced alpha-Gal A enzyme activity in Fabry fibroblast cells and Fabry mice tissues induced by serum from antibody positive patients with Fabry disease. In: Molecular Genetics and Metabolism. Band 94, Nummer 3, Juli 2008, S. 313–318, doi:10.1016/j.ymgme.2008.03.008. PMID 18456533.
- ↑ Lenders et al., J Allergy and Clin. Immunology, 2018.
- ↑ Maarten Arends, Marieke Biegstraaten u. a.: Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: an international cohort study. In: Journal of Medical Genetics. Band 55, 2018, S. 351, doi:10.1136/jmedgenet-2017-104863.
- ↑ Pharmazeutische Zeitung, Ausgabe 05 2016
- ↑ a b c S. D. James: Fabry Disease Patients Get Sicker as Drugs Go Overseas. In: abcNews.com, 30. August 2011
- ↑ C. A. Black: To march in or not to march in. In: Nature medicine. Band 17, Nummer 8, 2011, S. 921, doi:10.1038/nm.2440. PMID 21818082.
- ↑ rme: EMEA: Rationierung von Cerezyme und Fabrazyme. (Seite nicht mehr abrufbar, festgestellt im April 2019. Suche in Webarchiven) In: Aerzteblatt.de, 28. Juni 2009
- ↑ Supply shortages of Cerezyme and Fabrazyme – priority access for patients most in need of treatment recommended. (PDF; 35 kB) Pressemitteilung der EMEA vom 25. Juni 2009
- ↑ G. Schuh, C. Nußbaum u. a.: Umgang mit Know-how in internationalen FuE-Kooperationen. (PDF; 1,7 MB). Bundesministerium für Bildung und Forschung (Hrsg.), 2009, S. 21.
- ↑ a b E. Dolgin: NIH faces marching orders on orphan drug shortage. In: Nature medicine. Band 17, Nummer 5, Mai 2011, S. 522, doi:10.1038/nm0511-522b. PMID 21546953.
- ↑ M. Ratner: Fabry drug march-in denied. In: Nature biotechnology. Band 29, Nummer 97, 2011, S. 676, doi:10.1038/nbt0211-97b.
- ↑ F. S. Collins: Determination in the Case of Fabrazyme Manufactured by Genzyme Corporation. (PDF; 544 kB) Vom 1. Dezember 2010
- ↑ T. Staton: Genzyme apologizes for latest Fabrazyme delay. In: FiercePharma. 8. September 2011.
- ↑ T. Staton: Quality-control issue disrupts Fabrazyme flow. In: FiercePharma. 5. August 2011.
- ↑ a b Sanofi’s Genzyme starts shipping Fabry disease drug. In: Pharma Times. 2. März 2012, abgerufen am 30. April 2019 (englisch).
- ↑ a b Shire zieht Zulassungsantrag bei der FDA zurück. Ärzte Zeitung, 16. März 2012, abgerufen am 29. April 2019.
- ↑ Shire withdraws Replagal in USA as FDA wants more trials. Pharma Times, 15. März 2012, abgerufen am 29. April 2019.
- ↑ Timeline for Fabrazyme, Replagal*. (PDF) 14. Juli 2014, abgerufen am 29. April 2019.
- ↑ U. Fricke, U. Schwabe: Neue Arzneimittel 2007. In: U. Schwabe, D. Paffrath (Hrsg.): Arzneiverordnungs-Report 2008. Verlag Springer, 2008, S. 74. eingeschränkte Vorschau in der Google-Buchsuche
- ↑ B. Hoffmann, E. Mayatepek: Fabry disease-often seen, rarely diagnosed. In: Deutsches Ärzteblatt international. Band 106, Nummer 26, Juni 2009, S. 440–447, doi:10.3238/arztebl.2009.0440. PMID 19623315. PMC 270439 (freier Volltext). (Review).
- ↑ Eintrag EU/1/23/1724 im Unionsregister der europäischen Kommission, abgerufen am 10. Mai 2023.
- ↑ Novel Drug Approvals for 2023, FDA, abgerufen am 10. Mai 2023.
- ↑ Malte Lenders, Solvey Pollmann, Melina Terlinden, Eva Brand: Pre-existing anti-drug antibodies in Fabry disease show less affinity for pegunigalsidase alfa. In: . 2022, Band 26, S. 323–330. doi:10.1016/j.omtm.2022.07.009.
- ↑ Sam Kant, Mohamed G. Atta: Therapeutic advances in Fabry disease: The future awaits. In: . 2020, Band 131, S. 110779. doi:10.1016/j.biopha.2020.110779.
- ↑ Assessment Report Elfabrio, Ausschuss für Humanarzneimittel der EMA, 23. Februar 2023.
- ↑ Galafold. In: EMA Europa. 17. September 2018, abgerufen am 2. April 2019 (englisch).
- ↑ S. Ishii, H. H. Chang u. a.: Mutant alpha-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. In: The Biochemical journal. Band 406, Nummer 2, September 2007, S. 285–295, doi:10.1042/BJ20070479. PMID 17555407. PMC 1948963 (freier Volltext).
- ↑ S. Ishii, H. H. Chang u. a.: Preclinical efficacy and safety of 1-deoxygalactonojirimycin in mice for Fabry disease. In: The Journal of pharmacology and experimental therapeutics. Band 328, Nummer 3, März 2009, S. 723–731, doi:10.1124/jpet.108.149054. PMID 19106170.
- ↑ R. Hamanaka, T. Shinohara u. a.: Rescue of mutant alpha-galactosidase A in the endoplasmic reticulum by 1-deoxygalactonojirimycin leads to trafficking to lysosomes. In: Biochimica et biophysica acta. Band 1782, Nummer 6, Juni 2008, S. 408–413, doi:10.1016/j.bbadis.2008.03.001. PMID 18381081.
- ↑ a b S. Biastoff, B. Dräger: Calystegines. In: G. A. Cordell (Hrsg.): The Alkaloids: Chemistry and Biology. S. 91. eingeschränkte Vorschau in der Google-Buchsuche
- ↑ G. H. Yam, N. Bosshard u. a.: Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. In: American journal of physiology. Band 290, Nummer 4, April 2006, S. C1076–C1082, doi:10.1152/ajpcell.00426.2005. PMID 16531566.
- ↑ N. Asano, S. Ishii u. a.: In vitro inhibition and intracellular enhancement of lysosomal alpha-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. In: European Journal of Biochemistry. Band 267, Nummer 13, Juli 2000, S. 4179–4186. PMID 10866822.
- ↑ a b G. Andreotti, M. R. Guarracino u. a.: Prediction of the responsiveness to pharmacological chaperones: lysosomal human alpha-galactosidase, a case of study. In: Orphanet Journal of Rare Diseases. Band 5, 2010, S. 36, doi:10.1186/1750-1172-5-36. PMID 21138548. PMC 3016270 (freier Volltext). (Open Access)
- ↑ R. Khanna, R. Soska u. a.: The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. In: Molecular therapy. Band 18, Nummer 1, Januar 2010, S. 23–33, doi:10.1038/mt.2009.220. PMID 19773742. PMC 2839206 (freier Volltext).
- ↑ Klinische Studie (Phase III): Study of the Effects of Oral AT1001 (Migalastat Hydrochloride) in Patients With Fabry Disease bei Clinicaltrials.gov der NIH
- ↑ Entscheidung der Kommission vom 22-V-2006 über die Ausweisung des Arzneimittels „1-deoxygalactonojirimycin Hydrochlorid“ als Arzneimittel für seltene Leiden gemäß Verordnung (EG) Nr. 141/2000 des Europäischen Parlaments und des Rates. (PDF; 17 kB) (PDF) Vom 22. Mai 2006.
- ↑ S. H. Shin, S. Kluepfel-Stahl u. a.: Prediction of response of mutated alpha-galactosidase A to a pharmacological chaperone. In: Pharmacogenetics and genomics. Band 18, Nummer 9, September 2008, S. 773–780, doi:10.1097/FPC.0b013e32830500f4. PMID 18698230. PMC 2657085 (freier Volltext).
- ↑ T. Watt, A. P. Burlina u. a.: Agalsidase beta treatment is associated with improved quality of life in patients with Fabry disease: findings from the Fabry Registry. In: Genetics in medicine. Band 12, Nummer 11, November 2010, S. 703–712, doi:10.1097/GIM.0b013e3181f13a4a. PMID 20885332.
- ↑ M. R. Filling-Katz, H. F. Merrick u. a.: Carbamazepine in Fabry’s disease: effective analgesia with dose-dependent exacerbation of autonomic dysfunction. In: Neurology. Band 39, Nummer 4, April 1989, S. 598–600. PMID 2494569.
- ↑ National Institute for Health and Clinical Excellence: Neuropathic pain: The pharmacological management of neuropathic pain in adults in non-specialist settings (NICE clinical guideline 96). Centre for Clinical Practice at NICE; 2010.
- ↑ R. Baron, A. Binder, G. Wasner: Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. In: Lancet neurology. Band 9, Nummer 8, August 2010, S. 807–819, doi:10.1016/S1474-4422(10)70143-5. PMID 20650402. (Review).
- ↑ G. Cruccu, C. Sommer u. a.: EFNS guidelines on neuropathic pain assessment: revised 2009. In: European journal of neurology. Band 17, Nummer 8, August 2010, S. 1010–1018, doi:10.1111/j.1468-1331.2010.02969.x. PMID 20298428. (Review).
- ↑ C. E. Argoff, N. W. Barton u. a.: Gastrointestinal symptoms and delayed gastric emptying in Fabry’s disease: response to metoclopramide. In: Nuclear medicine communications. Band 19, Nummer 9, September 1998, S. 887–891. PMID 10581595.
- ↑ H. Tahir, L. L. Jackson, D. G. Warnock: Antiproteinuric therapy and fabry nephropathy: sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase-beta. In: JASN. Band 18, Nummer 9, September 2007, S. 2609–2617, doi:10.1681/ASN.2006121400. PMID 17656478.
- ↑ a b C. E. Stetter: In vivo Untersuchung des kardialen Stoffwechsels bei Morbus Fabry mittels 31Phosphor-MR-Spektroskopie. Dissertation. Julius-Maximilians-Universität zu Würzburg, 2007.
- ↑ I. B. Tomasic, M. C. Metcalf u. a.: Interconversion of the specificities of human lysosomal enzymes associated with Fabry and Schindler diseases. In: The Journal of biological chemistry. Band 285, Nummer 28, Juli 2010, S. 21560–21566, doi:10.1074/jbc.M110.118588. PMID 20444686. PMC 2898384 (freier Volltext).
- ↑ A. Abe, S. Gregory u. a.: Reduction of globotriaosylceramide in Fabry disease mice by substrate deprivation. In: The Journal of clinical investigation. Band 105, Nummer 11, Juni 2000, S. 1563–1571, doi:10.1172/JCI9711. PMID 10841515. PMC 300859 (freier Volltext).
- ↑ a b c T. Heare, N. J. Alp u. a.: Severe endothelial dysfunction in the aorta of a mouse model of Fabry disease; partial prevention by N-butyldeoxynojirimycin treatment. In: Journal of inherited metabolic disease. Band 30, Nummer 1, Februar 2007, S. 79–87, doi:10.1007/s10545-006-0473-y. PMID 17189993.
- ↑ EMA Europa: Zavesca. 17. September 2018, abgerufen am 2. April 2019 (englisch).
- ↑ G. M. Pastores, N. L. Barnett, E. H. Kolodny: An open-label, noncomparative study of miglustat in type I Gaucher disease: efficacy and tolerability over 24 months of treatment. In: Clinical therapeutics. Band 27, Nummer 8, August 2005, S. 1215–1227, doi:10.1016/j.clinthera.2005.08.004. PMID 16199246.
- ↑ C. E. Hollak, D. Hughes u. a.: Miglustat (Zavesca) in type 1 Gaucher disease: 5-year results of a post-authorisation safety surveillance programme. In: Pharmacoepidemiology and drug safety. Band 18, Nummer 9, September 2009, S. 770–777, doi:10.1002/pds.1779. PMID 19507165.
- ↑ a b T. Ohshima, R. Schiffmann u. a.: Aging accentuates and bone marrow transplantation ameliorates metabolic defects in Fabry disease mice. In: PNAS. Band 96, Nummer 11, Mai 1999, S. 6423–6427. PMID 10339603. PMC 26897 (freier Volltext).
- ↑ T. Takenaka, G. J. Murray u. a.: Long-term enzyme correction and lipid reduction in multiple organs of primary and secondary transplanted Fabry mice receiving transduced bone marrow cells. In: PNAS. Band 97, Nummer 13, Juni 2000, S. 7515–7520, doi:10.1073/pnas.120177997. PMID 10840053. PMC 16577 (freier Volltext).
- ↑ T. Yokoi, H. Kobayashi u. a.: Minimum requirement of donor cells to reduce the glycolipid storage following bone marrow transplantation in a murine model of Fabry disease. In: The journal of gene medicine. Band 13, Nummer 5, Mai 2011, S. 262–268, doi:10.1002/jgm.1566. PMID 21520359.
- ↑ S. H. Kim, P. D. Robbins: Retroviral vectors for gene therapy. In: J. A. Barranger, M. A. Cabrera-Salazar (Hrsg.): Lysosomal storage disorders. Verlag Springer, 2007, ISBN 978-0-387-70908-6, S. 53–63. eingeschränkte Vorschau in der Google-Buchsuche
- ↑ J. Park, G. J. Murray u. a.: Long-term correction of globotriaosylceramide storage in Fabry mice by recombinant adeno-associated virus-mediated gene transfer. In: PNAS. Band 100, Nummer 6, März 2003, S. 3450–3454, doi:10.1073/pnas.0537900100. PMID 12624185. PMC 152313 (freier Volltext).
- ↑ G. Qin, T. Takenaka u. a.: Preselective gene therapy for Fabry disease. In: PNAS. Band 98, Nummer 6, März 2001, S. 3428–3433, doi:10.1073/pnas.061020598. PMID 11248095. PMC 30670 (freier Volltext).
- ↑ J. E. Scherberich: Hereditäre Nierenerkrankungen. In: G. Steinbeck, G. Paumgartner: Therapie innerer Krankheiten. Verlag Springer, 2005, ISBN 3-540-23750-X, S. 576. eingeschränkte Vorschau in der Google-Buchsuche
- ↑ Y. A. Ioannou, K. M. Zeidner u. a.: Fabry disease: preclinical studies demonstrate the effectiveness of alpha-galactosidase A replacement in enzyme-deficient mice. In: American journal of human genetics. Band 68, Nummer 1, Januar 2001, S. 14–25, doi:10.1086/316953. PMID 11115376. PMC 1234907 (freier Volltext).
- ↑ M. E. Haskins, U. Giger, D. F. Patterson: Animal models of lysosomal storage diseases: their development and clinical relevance. In: A. Mehta, M. Beck, G. Sunder-Plassmann (Hrsg.): Fabry Disease: Perspectives from 5 Years of FOS. Kapitel 6, Oxford PharmaGenesis, 2006, ISBN 1-903539-03-X. PMID 21290677
- ↑ a b K. D. MacDermot, A. Holmes, A. H. Miners: Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. In: Journal of medical genetics. Band 38, Nummer 11, November 2001, S. 769–775. PMID 11732485. PMC 1734754 (freier Volltext).
- ↑ A. Mehta, J. T. Clarke u. a.: Natural course of Fabry disease: changing pattern of causes of death in FOS – Fabry Outcome Survey. In: Journal of medical genetics. Band 46, Nummer 8, August 2009, S. 548–552, doi:10.1136/jmg.2008.065904. PMID 19473999.
- ↑ a b c d e H. Fabry: An historical overview of Fabry disease. In: Journal of inherited metabolic disease. Band 24, Suppl 2, 2001, S. 3–7. PMID 11758677.
- ↑ a b c J. Fabry: Ein Beitrag zur Kenntnis der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae). In: Archiv für Dermatologie und Syphilis. Band 42, 1898, S. 187–200. doi:10.1007/BF01986897
- ↑ a b c T. Jansen, F. G. Bechara, P. Altmeyer: Angiokeratome – Klinik, Diagnostik und Therapie. TKT – Europe 5S, 2003, ISBN 3-00-011357-6.
- ↑ a b J. Fabry: Zur Klinik und Ätiologie des Angiokeratoma. In: Archiv für Dermatologie und Syphilis. Band 123, Nummer 2, 1916, S. 294–307.
- ↑ J. Fabry: Weiterer Beitrag zur Klinik des Angiokeratoma naeviforme (Naevus angiokeratosus). In: Dermatol Wochenschr. Band 90, 1930, S. 339–341.
- ↑ W. Anderson: A case of „Angeio-keratoma“. In: Br J Dermatol. Band 10, 1898, S. 113–117. doi:10.1111/j.1365-2133.1898.tb16317.x
- ↑ H. J. Wallace: Angiokeratoma corporis diffusum. In: Br J Dermatol. Band 70, 1968, S. 354–360. doi:10.1111/j.1365-2133.1958.tb13796.x PMID 13584689
- ↑ W. Cottle: Warty growths. In: St. Georges Hospital Reports. 9, 1877–1878, S. 753–762.
- ↑ J. Weicksel: Angiomatosis bzw. Angiokeratosis universalis (eine sehr seltene Haut- und Gefäßkrankheit). In: Dtsch Med Wochenschr. Band 51, 1925, S. 898–900.
- ↑ A. W. Pompen, M. Ruiter, H. J. Wyers: Angiokeratoma corporis diffusum (universale) Fabry, as a sign of an unknown internal disease; two autopsy reports. In: Acta medica Scandinavica. Band 128, Nummer 3, Juni 1947, S. 234–255. PMID 18897399.
- ↑ P. Reuter: Springer Großwörterbuch Medizin. Verlag Springer, 2004, ISBN 3-540-21352-X, S. 1186. eingeschränkte Vorschau in der Google-Buchsuche
- ↑ K. Scriba: Zur Pathogenese des Angiokeratoma corporis diffusum Fabry mit cardio-vasorenalem Symptomenkomplex. In: Verh Dtsch Ges Pathol. Band 34, 1950, S. 221–226.
- ↑ H. Hornbostel, K. Scriba: Zur Diagnostik des Angiokeratoma Fabry mit kardiovasorenalem Symptomenkomplex als Phosphatidspeicherkrankheit durch Probeexcision der Haut. In: Klinische Wochenschrift. Band 31, Nummer 3–4, Januar 1953, S. 68–69. PMID 13062573.
- ↑ J. R. Colley, D. L. Miller u. a.: The renal lesion in angiokeratoma corporis diffusum. In: British medical journal. Band 1, Nummer 5082, Mai 1958, S. 1266–1268. PMID 13536474. PMC 2028866 (freier Volltext).
- ↑ C. C. Sweeley, B. Klionsky: Fabry’s disease. Classification as sphingolipidosis and partial characterization of a novel glycolipid. In: The Journal of biological chemistry. Band 238, September 1963, S. 3148–3150. PMID 14081947.
- ↑ J. M. Opitz, F. C. Stiles u. a.: The Genetics of Angiokeratoma Corporis Diffusum (Fabry’s Disease) and Its Linkage Relations with the Xg Locus. In: American journal of human genetics. Band 17, Nummer 4, Juli 1965, S. 325–342. PMID 17948499. PMC 1932618 (freier Volltext).
- ↑ K. Hashimoto, B. G. Gross, W. F. Lever: Angiokeratoma Corporis Diffusum (Fabry). Histochemical and Electron Microscopic Studies of the Skin. In: The Journal of investigative dermatology. Band 44, Februar 1965, S. 119–128, doi:10.1038/jid.1965.22 PMID 14258908.
- ↑ M. Niemann: Der Einfluss langjähriger Enzymersatztherapie auf die Morphologie und Funktion des linken Ventrikels bei Patienten mit Morbus Fabry. Dissertation. Julius-Maximilians-Universität Würzburg, 2009.
- ↑ R. O. Brady, A. E. Gal u. a.: Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. In: The New England journal of medicine. Band 276, Nummer 21, Mai 1967, S. 1163–1167, doi:10.1056/NEJM196705252762101. PMID 6023233.
- ↑ J. A. Kint: Fabry’s disease: alpha-galactosidase deficiency. In: Science. Band 167, Nummer 922, Februar 1970, S. 1268–1269. PMID 5411915.
- ↑ J. W. Kusiak, J. M. Quirk, R. O. Brady: Purification and properties of the two major isozymes of alpha-galactosidase from human placenta. In: The Journal of biological chemistry. Band 253, Nummer 1, Januar 1978, S. 184–190. PMID 201618.
- ↑ R. J. Desnick, K. Y. Allen u. a.: Fabry’s disease: enzymatic diagnosis of hemizygotes and heterozygotes. Alpha-galactosidase activities in plasma, serum, urine, and leukocytes. In: The Journal of laboratory and clinical medicine. Band 81, Nummer 2, Februar 1973, S. 157–171. PMID 4683418.
- ↑ R. O. Brady, J. F. Tallman u. a.: Replacement therapy for inherited enzyme deficiency. Use of purified ceramidetrihexosidase in Fabry’s disease. In: The New England journal of medicine. Band 289, Nummer 1, Juli 1973, S. 9–14, doi:10.1056/NEJM197307052890103. PMID 4196713.
- ↑ R. J. Desnick, K. J. Dean u. a.: Enzyme therapy in Fabry disease: differential in vivo plasma clearance and metabolic effectiveness of plasma and splenic alpha-galactosidase A isozymes. In: PNAS. Band 76, Nummer 10, Oktober 1979, S. 5326–5330. PMID 228284. PMC 413135 (freier Volltext).
- ↑ C. A. Mapes, R. L. Anderson u. a.: Enzyme replacement in Fabry’s disease, an inborn error of metabolism. In: Science. Band 169, Nummer 949, September 1970, S. 987–989. PMID 4914726.
- ↑ T. Ohshima, G. J. Murray u. a.: alpha-Galactosidase A deficient mice: a model of Fabry disease. In: PNAS. Band 94, Nummer 6, März 1997, S. 2540–2544. PMID 9122231. PMC 20124 (freier Volltext).
- ↑ D. F. Bishop, D. H. Calhoun u. a.: Human alpha-galactosidase A: nucleotide sequence of a cDNA clone encoding the mature enzyme. In: PNAS. Band 83, Nummer 13, Juli 1986, S. 4859–4863. PMID 3014515. PMC 323842 (freier Volltext).
- ↑ R. Kornreich, R. J. Desnick, D. F. Bishop: Nucleotide sequence of the human alpha-galactosidase A gene. In: Nucleic Acids Research. Band 17, Nummer 8, April 1989, S. 3301–3302. PMID 2542896. PMC 317741 (freier Volltext).
- ↑ D. F. Bishop, R. Kornreich, R. J. Desnick: Structural organization of the human alpha-galactosidase A gene: further evidence for the absence of a 3' untranslated region. In: PNAS. Band 85, Nummer 11, Juni 1988, S. 3903–3907. PMID 2836863. PMC 280328 (freier Volltext).
- ↑ R. Schiffmann, G. J. Murray u. a.: Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. In: PNAS. Band 97, Nummer 1, Januar 2000, S. 365–370. PMID 10618424. PMC 26669 (freier Volltext).
- ↑ Agalsidase beta (Fabrazyme® Trockensubstanz; Genzyme GmbH). Abgerufen am 1. September 2011
- ↑ Agalsidase alfa (Replagal® Infusionslösung; TKT Europe). Abgerufen am 1. September 2011